

Key Points
Whole-exome sequencing and gene expression profiling reveal genetic driver alterations and elucidate pathway dependencies in PMBL.
Comparative analysis points to relevant differences to diffuse large B-cell lymphoma and highlights the pathological and molecular relatedness to cHL.

Abstract
Primary mediastinal large B-cell lymphoma (PMBL) represents a clinically and pathologically distinct subtype of large B-cell lymphomas. Furthermore, molecular studies, including global gene expression profiling, have provided evidence that PMBL is more closely related to classical Hodgkin lymphoma (cHL). Although targeted sequencing studies have revealed a number of mutations involved in PMBL pathogenesis, a comprehensive description of disease-associated genetic alterations and perturbed pathways is still lacking. Here, we performed whole-exome sequencing of 95 PMBL tumors to inform on oncogenic driver genes and recurrent copy number alterations. The integration of somatic gene mutations with gene expression signatures provides further insights into genotype–phenotype interrelation in PMBL. We identified highly recurrent oncogenic mutations in the Janus kinase-signal transducer and activator of transcription and nuclear factor κB pathways, and provide additional evidence of the importance of immune evasion in PMBL (CIITA, CD58, B2M, CD274, and PDCD1LG2). Our analyses highlight the interferon response factor (IRF) pathway as a putative novel hallmark with frequent alterations in multiple pathway members (IRF2BP2, IRF4, and IRF8). In addition, our integrative analysis illustrates the importance of JAK1, RELB, and EP300 mutations driving oncogenic signaling. The identified driver genes were significantly more frequently mutated in PMBL compared with diffuse large B-cell lymphoma, whereas only a limited number of genes were significantly different between PMBL and cHL, emphasizing the close relation between these entities. Our study, performed on a large cohort of PMBL, highlights the importance of distinctive genetic alterations for disease taxonomy with relevance for diagnostic evaluation and therapeutic decision-making.
Introduction
Over the past decade, a number of studies have unraveled the molecular and genetic heterogeneity of lymphoid malignancies, leading to the recognition of new entities and refinement of the existing taxonomy in the form of the World Health Organization (WHO) classification.1 In the broader context of the development of alternative treatment options, including the testing of novel drugs and/or combinations, coupled with refining a more personalized medicine approach, evidence is accumulating that disease classification plays an important role for patient risk stratification and therapeutic decision-making.
Large B-cell lymphomas account for >40% of B-cell non-Hodgkin lymphomas (NHL), representing the most common subtype worldwide.1 Diffuse large B-cell lymphoma (DLBCL), an aggressive cancer requiring immediate treatment, is the most common B-cell NHL and exemplifies the relevance of molecular subclassification and comprehensive genomic characterization for understanding fundamental disease biology and improving patient care.2-7 In contrast, primary mediastinal large B-cell lymphoma (PMBL) represents a relatively rare subtype, accounting for only 2% to 3% of B-cell NHL and 10% of large B-cell lymphomas. Its characteristic presentation features a predominant anterior mediastinal mass in young adults, with a higher prevalence in female subjects.1 Molecular studies in the past have provided evidence that PMBL truly typifies a distinct large B-cell lymphoma entity. In particular, gene expression profile analyses have shown that PMBL is more closely related to classical Hodgkin lymphoma (cHL)8,9 despite the fact that its morphological features are mostly reminiscent of DLBCL in terms of cell size, phenotype, and infiltration pattern.1,10 Although PMBL has been recognized as a distinct lymphoma entity in the WHO classification since 2008,1 diagnosis in most cases is still based on a clinico-pathological consensus that is not always well instituted and has a subjective component. This approach is in part due to the fact that gene expression profiling has not yet fully entered routine clinical practice, a problem that may be overcome by broader implementation of technologies using archival tissue specimens.11-13 Moreover, the correct identification of PMBL can be difficult because PMBL(-like) cases have been observed at non-mediastinal sites,14 and so-called mediastinal gray-zone lymphomas present another challenge in the spectrum of differential diagnoses.15-17
Since the first description of PMBL, a number of recurrent somatic gene mutations, copy number (CN) alterations, and frequently occurring structural genomic alterations have been identified. Constitutive activation of oncogenic signaling on the basis of mutations in the nuclear factor κB (NF-κB) and Janus kinase-signal transducer and activator of transcription (JAK-STAT) pathways is recognized as a hallmark of this disease.18,19 Moreover, key discoveries connecting genetic alterations with immune escape properties of lymphoma cells have highlighted the role of the tumor microenvironment in cancerogenesis20 and have spurred the implementation of immunotherapies, such as immune checkpoint blockade.21,22 Similar to cHL, PMBL harbors genomic aberrations leading to an immune privilege phenotype; namely, recurrent alterations targeting the class II transactivator CIITA,23,24 the master transcriptional regulator of major histocompatibility complex (MHC) class II, and numerical and structural genomic changes of the 9p24.1 locus25-28 (including the 2 programmed death 1-receptor ligands CD274 and PDCD1LG2) have been characterized as important aspects in PMBL pathogenesis.
Although targeted sequencing studies have revealed a number of key genetic lesions involved in PMBL lymphomagenesis, a comprehensive description of genetic alterations in a sizeable cohort of patients is still lacking. In the current study, we performed whole-exome sequencing of 95 PMBL tumors, most of which have also been molecularly defined by using the recently described Lymph3Cx assay.13 By systematically integrating mutational and gene expression profiles, we inform on oncogenic driver genes and recurrent CN alterations and describe new potential pathway dependencies. In addition, the intersection of driver genes found in related entities further substantiates the close relationship between PMBL and cHL.
Methods
Patient cohort and clinico-pathological characteristics
Patients were selected from the clinical database of the British Columbia Cancer, Centre for Lymphoid Cancer, based on a recorded (clinico-)pathological diagnosis of PMBL and clinical follow-up data. Specimens were retrieved from tissue archives of the Centre for Lymphoid Cancer or referring hospitals. In total, 118 biopsy specimens with the histopathological diagnosis of PMBL were identified that had all been previously reviewed by an expert panel of hematopathologists of the Lymphoma/Leukemia Molecular Profiling Project13 using criteria published in the 2008 WHO classification.1 Ninety-five of these cases were selected for this study based on availability of tissue, fresh-frozen cell suspensions, or tumor DNA. For 91 of 95 specimens, tumor content information was available and estimated to exceed 30% based on hematoxylin and eosin staining in 86 cases; 5 cases were macrodissected to enrich for tumor content or to exclude necrotic tumor areas. Tumor content information was not retrievable for 4 cases. Constitutional DNA from peripheral blood was available for 21 patients (in 12 cases, formalin-fixed, paraffin-embedded tissue-derived corresponding tumor DNA and in 9 cases, tumor DNA derived from cell suspensions was sequenced). The clinical characteristics are summarized in Table 1 and detailed information on study design, molecular classification, and available data per patient is given in supplemental Figure 1 (available on the Blood Web site).
Clinical characteristics of the PMBL cohort (N = 95)
| Clinical characteristic | n (%) |
|---|---|
| Sex | |
| Female | 54 (56.8) |
| Male | 41 (43.2) |
| Age, median (range), y | 34 (13-79) |
| Stage | |
| I/II | 70 (73.7) |
| III/IV | 24 (25.3) |
| NA | 1 (1) |
| Biopsy site | |
| Mediastinum | 59 (62.1) |
| Local or cervical lymph nodes, lung | 29 (30.5) |
| Inguinal lymph node, paraspinal L1 | 2 (2.1) |
| NA | 5 (5.3) |
| IPI group | |
| Low | 41 (43.1) |
| Intermediate | 39 (41.1) |
| High | 11 (11.6) |
| NA | 4 (4.2) |
| Bulky disease (>10 cm) | |
| Yes | 70 (73.7) |
| No | 24 (25.3) |
| NA | 1 (1) |
| Rituximab-containing therapy | |
| Yes | 59 (62.1) |
| No | 33 (34.7) |
| NA | 3 (3.2) |
| Clinical characteristic | n (%) |
|---|---|
| Sex | |
| Female | 54 (56.8) |
| Male | 41 (43.2) |
| Age, median (range), y | 34 (13-79) |
| Stage | |
| I/II | 70 (73.7) |
| III/IV | 24 (25.3) |
| NA | 1 (1) |
| Biopsy site | |
| Mediastinum | 59 (62.1) |
| Local or cervical lymph nodes, lung | 29 (30.5) |
| Inguinal lymph node, paraspinal L1 | 2 (2.1) |
| NA | 5 (5.3) |
| IPI group | |
| Low | 41 (43.1) |
| Intermediate | 39 (41.1) |
| High | 11 (11.6) |
| NA | 4 (4.2) |
| Bulky disease (>10 cm) | |
| Yes | 70 (73.7) |
| No | 24 (25.3) |
| NA | 1 (1) |
| Rituximab-containing therapy | |
| Yes | 59 (62.1) |
| No | 33 (34.7) |
| NA | 3 (3.2) |
IPI, International prognostic index; NA, not available.
This study was approved by the Research Ethics Board of the University of British Columbia and British Columbia Cancer (REB file H14-02304). Patients either provided written informed consent or a waiver of consent was in place for cases collected previously.
Whole-exome sequencing
DNA extraction was performed as previously described.24 DNA was derived from fresh-frozen cell suspensions in 32 cases and from formalin-fixed, paraffin-embedded tissue in 63 cases. Whole-exome sequencing was performed by using a targeted capture approach with the SureSelect Human All Exon V6+UTR bait (Agilent Technologies) followed by massively parallel sequencing of enriched fragments on the HiSeq 2500 platform (Illumina). Five libraries were pooled per lane, and a 125 bp paired-end mode was used. Tumor and normal DNA samples were sequenced to an average of 115X (standard deviation: 24X). All reads were aligned to the human reference genome (hg19) by using bwa-mem version 0.7.5a29 with optical and polymerase chain reaction duplicates removed by using the Picard tool (http://broadinstitute.github.io/picard/).
Identification of somatic single nucleotide variations and indels
For cases with paired normal DNA, somatic single nucleotide variation (SNV)/indel variants were identified by using the intersection of calls predicted by VarScan (version 2.3.6),30 Strelka (version 1.0.13),31 and MuTect (version 1.1.4),32 with a minimum variant allele frequency of 1% and 10 variant reads. For unpaired tumor specimens (n = 74), somatic SNV/indel variants were identified by using VarScan and required to have a minimum variant allele frequency of 5% and 10 variant reads. Putative germline variants were removed by using virtual normal correction.33 In addition, any variant associated with an identifier in dbSNP version 13734 was discarded. All variants were annotated by using SnpEff (version 4.2)35 and filtered for effects predicted to have a moderate or high impact at the protein level (ie, nonsynonymous, stop gained, splice site acceptor mutations).
MutSigCV analysis
The statistical significance of mutation frequency in each gene was determined by using the MutSigCV algorithm (http://software.broadinstitute.org/cancer/software/genepattern/modules/docs/MutSigCV).36 To accurately estimate the background mutation rate, synonymous mutations in addition to the full set of (nonsynonymous) somatic SNVs and indels identified through the paired SNV calling pipeline were analyzed. Because the default coverage file provided by MutSigCV may not properly represent the target space used by our capture bait, CovGen (https://github.com/tgen/CovGen) was used to provide an accurate estimate of coverage in the cohort. We used the default covariate table provided by MutSigCV, which contains the following variables for each gene: (1) global expression; (2) DNA replication time; and (3) HiC statistic. Due to our limited cohort size, P < .05 was used to obtain a list of candidate driver genes. From this list, only genes present in RefSeq were included for downstream analyses. Three genes (C21orf91, C11orf94, and C6orf226) had open reading frames with uncharacterized proteins and were therefore removed.
CN analysis
CN alterations for all tumor specimens were called by using the CNVkit37 with default thresholds (if log2 ≤ −4.3, CN = 0; if log2 ≤ −1, CN = 1; if log2 ≤ 0, CN = 2; if log2 ≤ 0.6, CN = 3) assuming a ploidy of 2 (further details are given in the supplemental Methods).
To identify relevant regions of CN alterations, the GISTIC (Genomic Identification of Significant Targets in Cancer) method described by Beroukhim et al38 was applied to the curated segmentation results (further details are given in the supplemental Methods).
Gene expression analysis and xseq
Gene expression data (DASL platform, HumanHT-12 v4.0 Expression BeadChip; Illumina) was available for 70 of 95 cases. RNA was extracted by using either the RNeasy Mini Kit or the AllPrep FFPE kit (Qiagen) and measured by using a NanoDrop spectrophotometer (Thermo Fisher Scientific). Nucleic acids were diluted to a final concentration of 100 ng/µl, and array hybridization was performed at The Hospital for Sick Children (Toronto, ON, Canada) using the HumanHT-12 v4.0 Expression BeadChip. The limma and beadarray R packages were used to analyze probe-level expression data. A normal-exponential convolution model was applied for background correction and normalization of the data, followed by log2 transformation and quality assessment. Probes below the detection score (ie, considered to be not expressed) in at least 3 arrays were removed (27 446 probes remained). One case of the sequencing cohort was identified as an outlier by both multidimensional scaling and principal component analysis, and was therefore removed. After exclusion of case PA012, a total of 68 cases were available for integrative analyses. Annotation of probes with quality and gene symbol information was performed by using the illuminaHumanv4.db probe annotation database (Illumina). Probes annotated as having “bad” quality or that had no gene match were removed. A total of 24 501 probes representing 17 027 genes remained, with 4659 of those being represented by >1 probe (ie, ambiguous transcript).
For both the cis- and trans-analysis (ie, mutations that affect gene expression of the same gene or a set of other genes, respectively), the xseq method was applied.39 Further details are given in the supplemental Methods.
Statistical analyses
Fisher’s exact test and, when appropriate, the χ2 test were used to assess whether differences in categorical variables (eg, frequency of mutated cases in different disease groups) were significant between groups. Statistical significance was defined as P < .05. All analyses were performed in R, the statistical programming language (R Core Team, 2017).
Results
The mutational landscape of PMBL identified by whole-exome sequencing
To characterize the full spectrum of coding sequence mutations in a large cohort of PMBL cases, we performed whole-exome sequencing in 95 patient-derived tumor specimens. Matched constitutional DNA was available for 21 cases. The clinical characteristics of these patients are described in Table 1. One tumor-normal pair was removed due to high similarity with the normal (likely due to insufficient tumor content in the frozen cell suspension), resulting in 20 paired cases that were used for downstream analysis.
By applying a robust bioinformatics pipeline integrating 3 variant callers (see "Methods" for details), 7200 somatic mutations were identified across the cohort of paired samples. Of those, 3120 were nonsynonymous (missense and nonsense), 1212 were synonymous, 2391 were nonexonic (3′- or 5′-untranslated region [UTR]), 73 were splice site alterations, and 404 were small indels (supplemental Figure 2A). After removal of silent and UTR variants, the median number of putatively protein-altering mutations per case was 155 (range, 44-741) in paired specimens (supplemental Figure 2B). A list of genes recurrently affected by UTR mutations is provided in supplemental Table 1.
Mutations in the unpaired specimens (n = 74) were filtered to exclude silent and UTR mutations, resulting in a median of 683 (range, 411-1227) protein-altering variants (SNVs and small indels) per case. The median number of variants in the unpaired specimens was significantly higher compared with the paired samples, attributable to the presence of private single nucleotide polymorphisms and platform noise that was not immediately filterable. In both paired and unpaired cases, SOCS1 (suppressor of cytokine signaling 1) was the most commonly mutated gene (64%). A complete list of 94 genes, which were mutated in at least 10% of all cases and 10% of paired cases (to reduce the number of likely artifacts), is provided in supplemental Table 2. We found frequently altered and previously identified18,40-43 genes involved in JAK-STAT signaling (STAT6 [signal transducer and activator of transcription 6], PTPN1 [protein tyrosine phosphatase nonreceptor type 1], IL4R [interleukin-4 receptor], JAK1, CISH [cytokine-inducible SH2-containing]), NF-κB signaling (TNFAIP3 [tumor necrosis factor, α-induced protein 3], NFKBIE [NFKB inhibitor ε], NFKB2 [NFKB subunit 2], and IKBKB [inhibitor of NFKB kinase subunit β]), and immune escape (CIITA, B2M [β2-microglobulin], and CD58 [cluster of differentiation 58]) (supplemental Figure 3A). Comparison of the median variant allele frequencies of these genes provides evidence that mutations in genes such as IL4R, FAT1 (FAT atypical cadherin 1), and EP300 (E1A binding protein P300) might occur early in PMBL pathogenesis, whereas those in CIITA, ACTB (actin β), and BTG1 (BTG antiproliferation factor 1) seem to be late events (supplemental Figure 3B). Interestingly, multiple genes involved in interferon response (IRF) were mutated in ∼10% of the tumors each. In particular, we found mutations in IRF1, IRF4, IRF8, and IRF2BP2 with identification of a hotspot mutation IRF4:c.295T>C, p.(Cys99Arg) in >50% of IRF4-mutated cases (supplemental Figure 4A). Overall, genetic alterations in these interferon response genes and related downstream targets were found in 52% of our cohort. Further analysis on a case-by-case level revealed a pattern reminiscent of mutual exclusivity, pointing toward a convergent role in PMBL pathogenesis (supplemental Figure 4B).
Oncogenic driver genes in PMBL
The tumor-normal pairs (n = 20) were used as a primary discovery set to identify potential genetic “drivers” (ie, gene mutations with high impact on disease pathogenesis) in PMBL, with the remaining cases serving as an internal validation set to ensure the reproducibility of findings, as well as an extension cohort to further characterize mutation frequencies and clinical correlates in the full set of 94 tumors. Application of MutSigCV36 to the distribution of somatic alterations in the discovery cohort revealed 50 putative driver genes that were recurrently and significantly mutated in PMBL (significance cutoff, P < .05) (supplemental Table 3). This gene set prominently included members of the JAK-STAT and NF-κB–signaling pathways (SOCS1, STAT6, PTPN1, IL4R, CISH, TNFAIP3, NFKBIE, and TRAF3 [TNF receptor associated factor 3]), genes known to be involved in immune escape (CIITA, CD58, and IL13RA), and interestingly several interferons and interferon-regulatory factors (IRF2BP2, IRF8, and IRF4). In line with these findings, gene ontology enrichment analysis depicted pathways related to B-cell activation, antigen processing and presentation, complement activation, IFN signaling, and Fc-γ receptor signaling (supplemental Figure 5A).
We next identified genetic alterations corresponding to these 50 candidate driver genes in all PMBL cases (n = 94); mutational frequencies, mutation types, and clinical/molecular features are displayed in Figure 1. Similarly, as described earlier, comparison of the median variant allele frequencies of these 50 candidate drivers suggests that mutations in genes such as IL4R, DDX3X (DEAD-box helicase 3), CD58, and NFKBIE occur early in PMBL pathogenesis, whereas CIITA, JUNB (JunB proto-oncogene, AP-1 transcription factor subunit), and KLF9 (Krüppel-like factor 9) are late events (supplemental Figure 5B). As an alternative approach, we used the OncodriveCLUST tool44 to identify potential driver genes in this dataset. A total of 29 genes were identified with only 5 genes having a false discovery rate (FDR) <0.1 (IRF4, EZH2 [enhancer of zeste homolog 2], STAT6, HIST1H3D, and ACTB) (supplemental Figure 6). Of note, these genes were also identified by MutSigCV.
![Figure 1. Putative driver genes in PMBL pathogenesis as identified by using MutSigCV. Each column in this plot represents an individual case (with mutation[s] in the displayed genes) of the final PMBL sequencing cohort (n = 94), separated into the discovery and extension cohort. Recurrently and significantly mutated genes (as identified by using the MutSigCV algorithm,36 P < .05) constitute individual rows and are sorted according to their mutational frequencies (in the form of absolute numbers of mutated cases as provided on the far right; note that for visualization purposes, of the fifty identified genes only genes mutated in ≥5% of the entire cohort are displayed). Tracks at the bottom of the plot provide information on molecular classification (using the Lymph3Cx assay13), sex, and biopsy site. Mutation types are color-coded as indicated in the key. ABC, activated B-cell; GCB, germinal center B-cell; N/A, not available.](https://ash.silverchair-cdn.com/ash/content_public/journal/blood/134/10/10.1182_blood.2019001126/3/m_bloodbld2019001126f1.png?Expires=1769096276&Signature=3Lf86qvgFnV61H5U5CGgum5Q9x8sIn6wl4Vc2bILoi5cAELgqOLG~--Yay83IGBCvse2G-5rH6teNCpVx2mZB6BijEfLmFgUd3SayIELsFsW9pCnEmrH667pww~89zzqEZxZcpoccMnM1uOsrwq1lClKrhDukGUJR00AddNOUlnTdY2vfmK7tnmPHRsxPoNaPBfVIjkeH~jSAycQxnrxoRgKUIstqMK0LV38GZYl1aeHQw57MEeiwQVseNQv5nrjOi2fMJoxUWiu0wPUC8RkAa8Lgpz4oPUqhSVfrVHgu3SbUSNR3sUdaRb2fWywRBVN9EPVv8sOv~d5RtVvhqosaw__&Key-Pair-Id=APKAIE5G5CRDK6RD3PGA)
Putative driver genes in PMBL pathogenesis as identified by using MutSigCV. Each column in this plot represents an individual case (with mutation[s] in the displayed genes) of the final PMBL sequencing cohort (n = 94), separated into the discovery and extension cohort. Recurrently and significantly mutated genes (as identified by using the MutSigCV algorithm,36 P < .05) constitute individual rows and are sorted according to their mutational frequencies (in the form of absolute numbers of mutated cases as provided on the far right; note that for visualization purposes, of the fifty identified genes only genes mutated in ≥5% of the entire cohort are displayed). Tracks at the bottom of the plot provide information on molecular classification (using the Lymph3Cx assay13 ), sex, and biopsy site. Mutation types are color-coded as indicated in the key. ABC, activated B-cell; GCB, germinal center B-cell; N/A, not available.
Putative driver genes in PMBL pathogenesis as identified by using MutSigCV. Each column in this plot represents an individual case (with mutation[s] in the displayed genes) of the final PMBL sequencing cohort (n = 94), separated into the discovery and extension cohort. Recurrently and significantly mutated genes (as identified by using the MutSigCV algorithm,36 P < .05) constitute individual rows and are sorted according to their mutational frequencies (in the form of absolute numbers of mutated cases as provided on the far right; note that for visualization purposes, of the fifty identified genes only genes mutated in ≥5% of the entire cohort are displayed). Tracks at the bottom of the plot provide information on molecular classification (using the Lymph3Cx assay13 ), sex, and biopsy site. Mutation types are color-coded as indicated in the key. ABC, activated B-cell; GCB, germinal center B-cell; N/A, not available.
Interestingly, when analyzing genetic alterations (including UTR and silent mutations) with regard to mutational signatures as defined by Alexandrov et al,45 we found an association of mutation patterns with aging, defective DNA mismatch repair, and AID/APOBEC activity (supplemental Figure 5C-E). Although the correlation with an “aging” signature seems counterintuitive in a patient population with a median age at diagnosis of 34 years, similar findings were reported in other cancers, including pediatric tumors.46 These findings might be reflective of the high proliferation and cell division count that is not necessarily linkable to the “chronological age” of the patient.
To investigate if genetic alterations in the identified candidate driver genes show patterns of co-occurrence or mutual exclusivity, we performed pairwise testing (Fisher’s exact test) followed by multiple test correction (Benjamini-Hochberg method). A number of significant correlations were identified between certain genetic lesions (Figure 2A), including 3 mutually exclusive pairs (SOCS1 and CXCR5 [CXC motif chemokine receptor 5]; GNA13 [guanine nucleotide-binding protein subunit α-13] and IL4R; STAT6 and CISH) (corrected P < .05) (Figure 2B). Examples for significantly co-occurring mutations (ACTB and IRF2BP2; ZFP36L1 and CISH) are depicted in supplemental Figure 7 (P < .05).
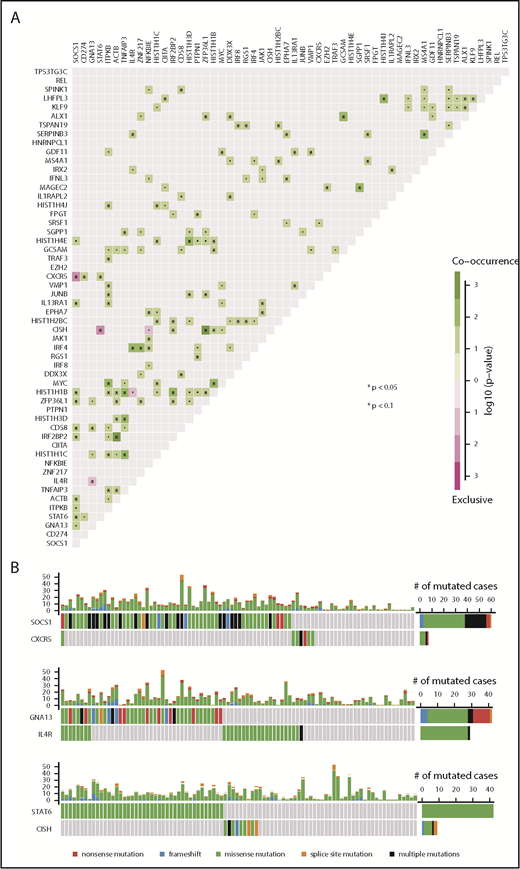
Mutational patterns and correlation of genetic alterations in PMBL. (A) Statistically significant mutual exclusivity or co-occurrence among the identified PMBL driver genes using pairwise Fisher’s exact test (multiple testing correction was performed according to the Benjamini-Hochberg method). (B) Highly significant examples of mutual exclusivity (SOCS1 and CXCR5, GNA13 and IL4R, STAT6 and CISH) are shown as pairwise comparisons. Each column represents a case, and mutated cases are highlighted in colors indicating specific mutation types as specified in the key. Absolute numbers of mutated cases are provided on the right, and the frequency plot on top of each subpanel provides the number of mutations in driver genes per case.
Mutational patterns and correlation of genetic alterations in PMBL. (A) Statistically significant mutual exclusivity or co-occurrence among the identified PMBL driver genes using pairwise Fisher’s exact test (multiple testing correction was performed according to the Benjamini-Hochberg method). (B) Highly significant examples of mutual exclusivity (SOCS1 and CXCR5, GNA13 and IL4R, STAT6 and CISH) are shown as pairwise comparisons. Each column represents a case, and mutated cases are highlighted in colors indicating specific mutation types as specified in the key. Absolute numbers of mutated cases are provided on the right, and the frequency plot on top of each subpanel provides the number of mutations in driver genes per case.
Recurrent CN alterations in PMBL cooperate with mutations in pathways related to immune evasion and oncogenic signaling
In addition to SNVs and indels, we characterized CN alterations across all 94 exomes using CNVkit,37 a tool that utilizes both on- and off-target coverage to infer CN evenly across the genome (supplemental Figure 8). GISTIC38 was then applied to identify regions of statistically significant gains or losses (Figure 3A). Consistent with earlier literature, the most significant regions of CN gain were located on 9p (CD274 and PDCD1LG2; 72% of tumors showing CN gain) (Figure 3B), 2p (REL; 36%), 6p (47%), and 11q (40%). Although CN losses are poorly characterized in PMBL, we found that the most frequent regions of loss identified in our study (chromosomes 10q, 8p, 1p, and 7p; all >30%) were consistent with those identified in the study by Wessendorf et al,47 which is the largest PMBL genome-wide CN study performed to date. Details on chromosomal location, including genes and statistics of significant regions of deletions and gains/amplifications, are provided in supplemental Table 4, and the distribution of these CN alterations across the PMBL cohort is shown in supplemental Figure 9B.
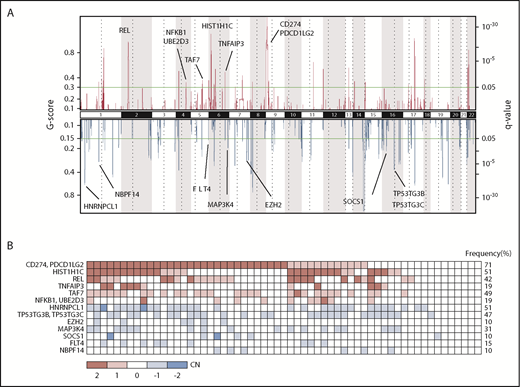
Recurrent CN alterations in PMBL. (A) Significant CN gains (red) and losses (blue) as inferred from whole-exome sequencing data using GISTIC 2.0. Potential candidate genes are highlighted next to their respective genomic locus. (B) Frequency plot of genes displaying significant CN alterations across the cohort. Red indicates CN gain, and blue indicates CN loss, where values are derived from GISTIC and indicate copy-number level per gene: −2 = homozygous deletion; −1 = heterozygous deletion; 0 = CN unchanged; 1 = low-level gain; 2 = high-level gain/amplification. Genes were sorted according to gain/deletion and then per q value as calculated by using GISTIC.
Recurrent CN alterations in PMBL. (A) Significant CN gains (red) and losses (blue) as inferred from whole-exome sequencing data using GISTIC 2.0. Potential candidate genes are highlighted next to their respective genomic locus. (B) Frequency plot of genes displaying significant CN alterations across the cohort. Red indicates CN gain, and blue indicates CN loss, where values are derived from GISTIC and indicate copy-number level per gene: −2 = homozygous deletion; −1 = heterozygous deletion; 0 = CN unchanged; 1 = low-level gain; 2 = high-level gain/amplification. Genes were sorted according to gain/deletion and then per q value as calculated by using GISTIC.
Contrasting mutational landscapes to related pathological entities
To define more clearly the commonalities and differences between PMBL and related pathological entities (ie, DLBCL and cHL), we compared the mutational driver genes in PMBL as described earlier vs 2 large cohorts of DLBCL4,48 (n = 304 and n = 1001, respectively) and the largest cHL exome cohort studied to date (n = 34).49 Interestingly, PMBL driver genes mutated in ≥3 tumors exhibited dramatically lower mutational frequencies in the cohort of patients with DLBCL reported by Reddy et al48 (Figure 4A). When we compared mutational frequencies in a panel of genes, representing the driver genes in DLBCL as reported by Chapuy et al4 (supplemental Figure 10), we could clearly show that, with the exception of EZH2, the frequencies of mutations in all these genes were statistically significantly different (P < .05) between PMBL and DLBCL (Figure 4B). In contrast, frequencies of recurrently mutated genes in cHL trended similarly to corresponding frequencies in the PMBL cohort, with only 3 genes (IGLL5, ITPKB, and NUP214) displaying a statistically significant difference (P < .05) (Figure 4C).

Comparison of mutational frequencies between histopathological/pathogenetically related entities. (A) Comparison of mutation frequencies for putative PMBL driver genes identified by MutSigCV between our PMBL cohort and the DLBCL cohort published by Reddy et al48 (genes were required to be mutated in at least 3 cases). (B) Comparison of mutation frequencies in DLBCL driver genes as identified in Chapuy et al4 between our PMBL cohort and the DLBCL cohorts published by Reddy et al and Chapuy et al; EZH2 is marked by an asterisk in panels A and B, as the only gene with no significant difference in terms of mutational frequency between DLBCL and PMBL. (C) Mutational frequencies in cHL and PMBL are comparable for genes shown to be recurrently mutated in cHL as defined by Tiacci et al.49 Statistically significant differences in mutation frequency between the two cohorts are marked with an asterisk. For all comparisons, silent as well as 5′- and 3′-UTR mutations were excluded.
Comparison of mutational frequencies between histopathological/pathogenetically related entities. (A) Comparison of mutation frequencies for putative PMBL driver genes identified by MutSigCV between our PMBL cohort and the DLBCL cohort published by Reddy et al48 (genes were required to be mutated in at least 3 cases). (B) Comparison of mutation frequencies in DLBCL driver genes as identified in Chapuy et al4 between our PMBL cohort and the DLBCL cohorts published by Reddy et al and Chapuy et al; EZH2 is marked by an asterisk in panels A and B, as the only gene with no significant difference in terms of mutational frequency between DLBCL and PMBL. (C) Mutational frequencies in cHL and PMBL are comparable for genes shown to be recurrently mutated in cHL as defined by Tiacci et al.49 Statistically significant differences in mutation frequency between the two cohorts are marked with an asterisk. For all comparisons, silent as well as 5′- and 3′-UTR mutations were excluded.
To better understand potential contrasting genomic underpinnings in tumors reflecting the molecular spectrum of PMBL and DLBCL, we next compared mutational profiles in our PMBL cohort based on molecular classification according to the Lymph3Cx assay.13 Interestingly, by contrasting mutational frequencies in bona fide PMBL (as assigned by the molecular classifier, n = 73) vs the cohort comprising molecularly defined DLBCL (n = 5) and tumors placed in the “uncertain” category (n = 12), we identified IL4R as being significantly enriched in the molecular PMBL group (with no mutations found in the other groups); TNFRSF14 was only found to be mutated in molecularly defined non-PMBL tumors (supplemental Figure 11A-C). Of note, when we analyzed the tumors with available gene expression data (DASL platform) based on the genes included in the recently published molecular classifier,13 we obtained a similar pattern, with the molecular DLBCLs forming a separate cluster, pointing to the stability of the gene expression–based model (supplemental Figure 12).
Integrative analysis
We next performed an integrated mutation–gene expression analysis using xseq39 to ascertain the impact of recurrent mutations on transcriptional profiles. Specifically, the approach aims at assessing the likely association of present mutations with observed deviations from neutral expression measurements in the same tumor. Examining the “cis-effect” of loss-of-function mutations (frameshift, nonsense, and splice site mutations) on gene expression, NBPF1 (neuroblastoma breakpoint family, member 1), mutated in 30 tumors, was identified with high certainty [P(D) = .9942] to have an influence on gene expression in its mutated form (Figure 5A), with 3 other genes (FRG1, STAT6, and ZNF681) having considerable “cis-effects” on gene expression but with probability levels below the recommended cutoff [P(D) < .8]. In contrast, a number of mutated genes exhibited “trans-effects” on gene expression with high-confidence probabilities (P(D) > 0.8). The distribution of mutations in those genes across the sequencing cohort with available gene expression data (n = 68) revealed alterations in at least 1 of these 15 genes on an individual case level (Figure 5B). Most prominently, we identified among the significantly trans-influencing genes, JAK1 and RELB, members of the JAK-STAT and NF-κB pathways, respectively. Both pathways are known activated oncogenic pathways in PMBL, although the importance of JAK1 and RELB mutations as drivers of differential expression programs has not been described before. The respective gene association networks with functionally enriched pathway members are displayed in Figure 5C and 5D, respectively. EP300, a histone acetyltransferase that functions as a coactivator of many transcription factors to regulate gene transcription, plays an important role in hematopoiesis and has been found mutated in other hematological malignancies.50,51 Figure 5E displays the gene association network and indicates quantitative changes in gene expression of functionally enriched pathway members.
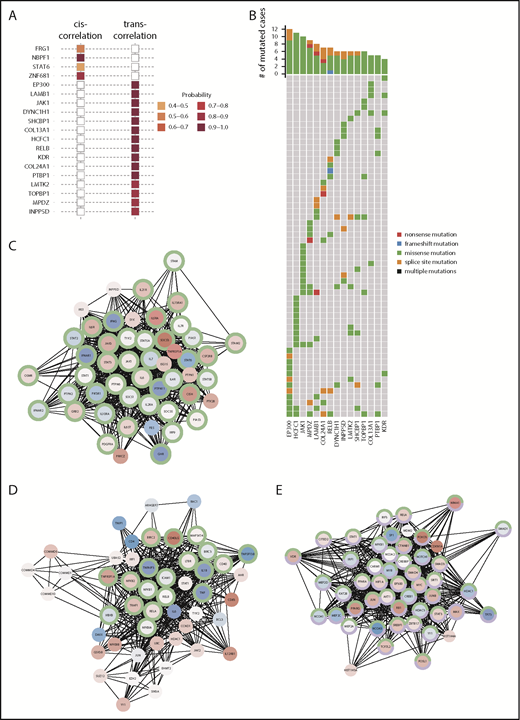
Impact of recurrent mutations on the transcriptome. (A) Plot displaying sets of genes identified as showing significant cis- or trans-effects on gene expression based on xseq analysis. (B) Oncoplot showing the distribution of mutations in genes with a trans-effect across the entire cohort. Gene association networks for significant trans-influencing genes exemplified by (C) JAK1, (D) RELB, and (E) EP300 based on xseq analysis. Nodes represent genes within the interaction network, and edges are interactions between genes based on the global influence network (see "Methods" for details). Nodes are colored by average log2 normalized expression across patients whose tumors harbor mutations, with red indicating high expression and blue indicating low expression. Genes with green borders in the network are functionally enriched for the JAK-STAT signaling pathway (panel C, FDR, P = 4.44E-72) or the NF-κB signaling pathway (panel D, FDR, P = 8.13E-27), respectively. In panel E, the green and purple borders highlight genes enriched in pathways related to epigenetic regulation, specifically the regulation of transcription (FDR, P = 8.3E-47) and the regulation of nucleic acid–templated transcription (FDR, P = 4.83E-43), respectively.
Impact of recurrent mutations on the transcriptome. (A) Plot displaying sets of genes identified as showing significant cis- or trans-effects on gene expression based on xseq analysis. (B) Oncoplot showing the distribution of mutations in genes with a trans-effect across the entire cohort. Gene association networks for significant trans-influencing genes exemplified by (C) JAK1, (D) RELB, and (E) EP300 based on xseq analysis. Nodes represent genes within the interaction network, and edges are interactions between genes based on the global influence network (see "Methods" for details). Nodes are colored by average log2 normalized expression across patients whose tumors harbor mutations, with red indicating high expression and blue indicating low expression. Genes with green borders in the network are functionally enriched for the JAK-STAT signaling pathway (panel C, FDR, P = 4.44E-72) or the NF-κB signaling pathway (panel D, FDR, P = 8.13E-27), respectively. In panel E, the green and purple borders highlight genes enriched in pathways related to epigenetic regulation, specifically the regulation of transcription (FDR, P = 8.3E-47) and the regulation of nucleic acid–templated transcription (FDR, P = 4.83E-43), respectively.
Discussion
The current article reports on whole-exome sequencing data obtained in a cohort of 94 PMBL tumors. This is, to our knowledge, the largest cohort for this entity reported to date, with previous studies limited by lower patient numbers and/or usage of only targeted sequencing approaches.40,52 In addition, the vast majority of lymphomas included in this study underwent rigorous pathology panel review and were molecularly classified by using the recently published Lymph3Cx assay.13 Using MutSigCV, we describe a set of 50 putative driver genes with clear evidence of somatic mutation status in a considerable proportion of tumors. Given the relatively small sample size of our discovery cohort, we selected a P value cutoff of .05, representing a stringency level that balances biological discovery potential with the chance of false-positive findings. As expected from previous studies reporting single gene alterations, mutations affecting various members of the JAK-STAT and NF-κB pathways are highly enriched in PMBL, with SOCS1 being the most frequently altered gene in our cohort. In concordance with the recent studies published by Dubois et al40 and Mansouri et al,52 we also found ITPKB, MFHAS1 (malignant fibrous histiocytoma amplified sequence 1), XPO1 (exportin 1), and NFKBIE to be frequently mutated in PMBL. A summary of the key findings of our study confirming and textualizing previous findings, as well as discovering novel disease biology, is presented in supplemental Figure 13.
Another prominent aspect of PMBL pathophysiology is the immune escape phenotype in this disease.20 Consistent with this, we discovered recurrent genomic alterations in genes involved in MHC expression and MHC complex formation/stability (B2M and CIITA), interaction with NK cell subsets (CD58), and alteration of T cell–mediated immune response (CD274 and PDCD1LG2), which have been, in part, extensively characterized before and were also recently reported in a series of 37 PMBL cases.23,24,27,53 In addition to known recurrent aberrations, we have observed mutations in genes involved in interferon signaling in one half of the tumors (IRF2BP2 in 22% of cases, IRF8 in 12%, IRF4 in 11%, and CISH in 10%). The transcription factors IRF4 and IRF8 are known to play an important role in B-cell/plasma cell development as well as in the germinal center response in a mutually antagonistic manner.54,55 Interestingly, we found in 6 (54.5%) of 11 cases, a hotspot mutation IRF4:c.295T>C, p.(Cys99Arg) affecting the DNA-binding domain of IRF4. This specific nucleotide change has been previously observed in activated B-cell–like DLBCL,40 although at lower relative frequencies compared with our study. To what extent this mutation constitutes IRF4-addiction in PMBL and a potential therapeutically targetable vulnerability needs to be determined in functional studies. Moreover, the interactions with other pathways known to play important roles in PMBL pathogenesis (eg, the JAK-STAT pathway) will need to be elucidated in the future.
By comparing mutational frequencies in PMBL and related pathological entities (DLBCL and cHL), we provide further evidence that PMBL is distinct from DLBCL not only on the gene expression level but also in terms of the mutational landscape and putative driver genes. Conversely, the majority of recurrently mutated genes in cHL are also frequently altered in PMBL, further establishing the pathophysiological relatedness of these 2 lymphoma entities.8,9
Our study also yielded specific genes (JAK1, RELB, and EP300) with disrupted expression triggered by genomic mutations in either the coding or the regulatory space. These results imply that mutated regulatory components of the genome contribute substantially to cancer pathways and show the importance of integrative analysis approaches to enhance biological interpretation of the PMBL mutational landscape and other cancers.
Recent publications in DLBCL4,5 suggest that mutational profiles, either in isolation or in synergy with cell-of-origin–based classification (activated B-cell– vs germinal center B-cell–like DLBCL) and standard diagnostic tools such as fluorescence in situ hybridization or immunohistochemistry, can aid in more precise classification of aggressive B-cell lymphomas. Our finding of a distinct mutational landscape in PMBL, in contrast to DLBCL, and select mutations (eg, in IL4R and TNFRSF14) cosegregating with molecular signatures strongly suggest that mutational profiling should be evaluated to establish a new taxonomy for aggressive lymphomas and tested for clinical validity and utility alongside clinical trials and modern treatments of PMBL and related entities.
The publication costs of this article were defrayed in part by page charge payment. Therefore, and solely to indicate this fact, this article is hereby marked “advertisement” in accordance with 18 USC section 1734.
The data reported in this article have been deposited in the Gene Expression Omnibus database (accession number GSE134511). Exomes are uploaded to the European Genome-Phenome Archive (accession number EGAS00001003746).
The online version of this article contains a data supplement.
Acknowledgments
The authors are grateful to J. Ding for technical support implementing the xseq tool. The authors also thank the pathology panel members of the Lymphoma/Leukemia Molecular Profiling Project for central pathology review of all PMBL cases used in this study.
This work was supported by generous funding from the Terry Fox Research Institute through a Program Project grant (1023 and 1061, C. Steidl, J.C., and M.M.) and a Canadian Institutes of Health Research Foundation grant to C. Steidl. C. Steidl is supported by a Michael Smith Foundation for Health Research career investigator award and a Canadian Institutes of Health Research new investigator award. A. Mottok was supported by postdoctoral and research fellowships from the German Cancer Aid (Mildred-Scheel-Foundation), the Michael Smith Foundation for Health Research (MSFHR), and Lymphoma Canada, and currently receives funding through the Clinician Scientist Program at the Medical Faculty, University of Ulm. E.V. is supported by a Michael Smith Foundation for Health Research trainee award.
Authorship
Contribution: A. Mottok, S.S.H., E.A.C., and C. Steidl designed the study; A. Mottok, S.S.H., E.A.C., B.W., A.T., B.M., H.N., C.R., and S.B.-N. performed research; A. Mottok, S.S.H., L.C.C., C.R., E.V., C. Sarkozy, A. Mungall, M.A.M., R.S., and C. Steidl interpreted data; R.D.G., J.M.C., D.W.S., and K.J.S. provided clinical data and/or performed pathology review; A. Mottok, S.S.H., and C. Steidl wrote the manuscript; and all authors read and approved the final manuscript.
Conflict-of-interest disclosure: A. Mottok, D.W.S., and C. Steidl are named inventors on a patent filed by the National Cancer Institute “Methods for determining lymphoma type.” K.J.S. has received honoraria from and consulted for Seattle Genetics, Bristol-Myers Squibb, Merck, Verastem, and AbbVie; has consulted for Servier; received honoraria from Takeda; and received institutional research funding from Roche. D.W.S. has served as a consultant for Janssen and Celgene; received research funding from Roche/Genentech, Janssen, and NanoString Technologies; received travel funding from Celgene; and is a named inventor on patents, including one licensed to NanoString Technologies (institution). C. Steidl has served as a consultant for Seattle Genetics, Roche, Bayer, and Curis; and has received research funding from Bristol-Myers Squibb and Tioma. The remaining authors declare no competing financial interests.
Correspondence: Christian Steidl, Department of Lymphoid Cancer Research, British Columbia Cancer, 675 West 10th Ave, Vancouver, BC V5Z 1L3, Canada; e-mail: csteidl@bccancer.bc.ca.

This feature is available to Subscribers Only
Sign In or Create an Account Close Modal