

Key Points
Zanubrutinib is a potent and selective BTK inhibitor with greater selectivity and potentially fewer off-target effects than ibrutinib.
In this study, zanubrutinib was tolerable and demonstrated encouraging activity in relapsed/refractory or treatment-naive CLL patients.

Abstract
Zanubrutinib is a potent and highly selective inhibitor of Bruton tyrosine kinase (BTK). In this first-in-human, open-label, multicenter, phase 1 study, patients in part 1 (3 + 3 dose escalation) had relapsed/refractory B-cell malignancies and received zanubrutinib 40, 80, 160, or 320 mg once daily or 160 mg twice daily. Part 2 (expansion) consisted of disease-specific cohorts, including treatment-naive or relapsed/refractory chronic lymphocytic leukemia/small lymphocytic lymphoma (CLL/SLL). The primary end points were safety and tolerability, and definition of the maximum tolerated dose (part 1). Additional end points included pharmacokinetics/pharmacodynamics and preliminary efficacy. Reported herein are results from 144 patients enrolled in the dose-finding and CLL/SLL cohorts. No dose-limiting toxicities occurred in dose escalation. Median BTK occupancy in peripheral blood mononuclear cells was >95% at all doses. Sustained complete (>95%) BTK occupancy in lymph node biopsy specimens was more frequent with 160 mg twice daily than 320 mg once daily (89% vs 50%; P = .0342). Consequently, 160 mg twice daily was selected for further investigation. With median follow-up of 13.7 months (range, 0.4-30.5 months), 89 CLL/SLL patients (94.7%) remain on study. Most toxicities were grade 1/2; neutropenia was the only grade 3/4 toxicity observed in >2 patients. One patient experienced a grade 3 subcutaneous hemorrhage. Among 78 efficacy-evaluable CLL/SLL patients, the overall response rate was 96.2% (95% confidence interval, 89.2-99.2). Estimated progression-free survival at 12 months was 100%. Zanubrutinib demonstrated encouraging activity in CLL/SLL patients, with a low incidence of major toxicities. This trial was registered at www.clinicaltrials.gov as #NCT02343120.
Introduction
The B-cell receptor signaling pathway is not only essential for normal B-cell development but is also implicated in the survival and proliferation of malignant B cells.1-3 Inhibition of B-cell receptor signaling has recently been established as an effective approach for management of B-cell malignancies.4 Bruton tyrosine kinase (BTK) is a key component of the B-cell receptor signaling pathway, and the first-generation BTK inhibitor, ibrutinib, has become a standard of care in frontline and previously treated chronic lymphocytic leukemia/small lymphocytic lymphoma (CLL/SLL), previously treated mantle cell lymphoma (MCL), and Waldenström macroglobulinemia (WM).5-10
Zanubrutinib (BGB-3111) is a highly specific next-generation BTK inhibitor with favorable oral bioavailability, as shown in preclinical studies.11-13 Compared with ibrutinib, zanubrutinib has shown greater selectivity for BTK and fewer off-target effects in multiple in vitro enzymatic and cell-based assays (supplemental Table 1, available on the Blood Web site).13 Zanubrutinib, ibrutinib, and other active BTK inhibitors covalently bind cysteine 481 in the adenosine triphosphate–binding pocket of BTK, and display varying affinities (depending on specificity of the individual drug) for related and unrelated adenosine triphosphate–binding kinases that contain a sterically available cysteine at this position, including epidermal growth factor receptor (EGFR), human EGFR-2 (HER2), human EGFR-4 (HER4), interleukin-2–inducible T-cell kinase (ITK), bone marrow tyrosine kinase gene in chromosome X (BMX), JAK2, TEC, and B-lymphocyte kinase (BLK).3,12,14,15 Off-target inhibition likely contributes to the toxicities reported in patients treated with ibrutinib, such as diarrhea and rash (toxicities associated with EGFR inhibition),5-7 bleeding or bruising,16,17 and atrial fibrillation,18,19 and those that are not seen in patients with congenital X-linked agammaglobulinemia due to germline mutations in the BTK gene; a more specific BTK inhibitor may have fewer toxicities.
Based on promising preclinical data, we conducted a phase 1 study of zanubrutinib to evaluate its safety, pharmacokinetic, and pharmacodynamic properties. Herein, we report results in patients with various relapsed or refractory B-cell malignancies and preliminary safety and efficacy results in patients with treatment-naive or relapsed/refractory CLL/SLL.
Patients and methods
Study design and participants
This multicenter, phase 1, first-in-human study of zanubrutinib in patients with B-cell malignancies comprises 2 parts: dose escalation (part 1) and cohort expansion (part 2). Part 1 evaluated the safety, pharmacokinetics, and pharmacodynamics (BTK occupancy in peripheral blood mononuclear cells [PBMCs]) in patients with relapsed/refractory B-cell malignancies who had received at least 1 prior therapy, with no therapy of higher priority available in the assessment of the investigator. Part 2 characterized the safety and preliminary efficacy of zanubrutinib in multiple cohorts of patients with B-cell malignancies. One cohort (cohort 2a) enrolled patients with mixed histologies of relapsed/refractory B-cell malignancies (see supplemental Figure 1, available on the Blood Web site). These patients underwent pharmacodynamic evaluations consisting of evaluation of pretreatment and on-treatment lymph node biopsy specimens and PBMC assessments for quantitation of BTK occupancy. Other cohorts were disease-specific, for example, CLL/SLL, MCL, and WM. Protocol amendment version 5 provided for the inclusion of patients with CLL/SLL who had not received prior treatment of their disease. This provision was based on the acceptable safety profile of zanubrutinib observed in part 1 (dose escalation) and early part 2 (dose expansion) of this study, as well as emerging data with ibrutinib and acalabrutinib demonstrating that BTK inhibition is safe and active, including in previously untreated patients.5
Eligible patients were at least 18 years old, had an Eastern Cooperative Oncology Group performance status of 0 to 2, and adequate organ function and peripheral blood counts (absolute neutrophil count, ≥1.0 × 109/L; platelet count, ≥50 × 109/L). Key exclusion criteria included central nervous system disease, previous exposure to a BTK inhibitor, or a requirement for concurrent strong cytochrome P450 3A (CYP3A) inhibitors or inducers. Additional eligibility criteria are included in the supplemental Methods. All patients provided written informed consent before enrollment, and institutional review boards and independent ethics committees reviewed and approved the study protocol.
Procedures
Part 1
In part 1, patients received daily zanubrutinib until disease progression or intolerable toxicity, with dose escalation according to a modified “3 + 3” design. The dose-limiting toxicity assessment window was 21 days after first dose. A safety monitoring committee was established to determine dose levels and schedules for dose escalation, using data from previous dose cohorts. Dose-limiting toxicities were defined as: treatment-related grade 4 neutropenia lasting >7 days, grade ≥3 febrile neutropenia of any duration, grade 4 thrombocytopenia or grade ≥3 thrombocytopenia associated with bleeding, any nonhematologic grade ≥3 event, any grade ≥2 toxicity requiring dose modification or ≥1 week treatment delay, or any toxicity that required study drug discontinuation. Patients tolerating their assigned dose were permitted to escalate to a higher dose if it was deemed safe in a subsequent cohort and after sponsor review. If no maximum tolerated dose was identified, decisions as to the dose and schedule(s) to be evaluated in part 2 were to be based on pharmacokinetic data, assessments of BTK inhibition in PBMCs, safety, and preliminary efficacy.
Mouse pharmacokinetic/pharmacodynamic models and human pharmacokinetic data predicted that zanubrutinib dosed at 80 mg per day would achieve complete BTK inhibition in PBMCs. In an attempt to achieve the anticipated therapeutic dose with ≤4 dose levels, 40 mg per day was selected as the starting dose for dose escalation with a planned maximum of 320 mg per day as either a single or split daily dose (160 mg twice daily).
Part 2
Patients enrolled in part 2 were to receive zanubrutinib at the maximum tolerated dose or maximally administered dose until disease progression, loss of clinical benefit, or intolerable toxicity. Continuous safety evaluation was performed by the sponsor and study investigators. Patients enrolled in cohort 2a provided consent to undergo paired lymph node biopsies for quantitation of BTK occupancy; biopsies were optional for patients enrolled in other part 2 cohorts. Safety and efficacy results from CLL/SLL patient cohorts (either treatment-naive or relapsed/refractory disease) in part 2 are also presented.
Pharmacokinetic and pharmacodynamic assessments: part 1 and cohort 2a patients
Blood samples for pharmacokinetic analyses were collected predose on days 1, 2, and 3 of week 1; day 1 of weeks 2, 5, and 9; and up to 8 hours postdose on day 1 of weeks 1 and 2. Zanubrutinib plasma concentrations were measured using a validated high-performance liquid chromatography tandem mass spectrometry assay with a lower limit of quantitation for zanubrutinib of 1.00 ng/mL. Pharmacokinetic parameters, including maximal plasma concentration (Cmax) and area under the concentration-time curve, were estimated using noncompartmental methods and were computed using Phoenix WinNonlin, version 7.0 (Certara).
Blood samples for quantitation of BTK occupancy were collected predose on days 1, 2, and 3 of week 1, on day 1 of week 2, and 4 hours postdose on day 1 of week 1. For quantitation of BTK occupancy in nodal tissues, patients enrolled on cohort 2a with accessible tissue underwent lymph node biopsies (core needle biopsy in most cases) at screening and predose (ie, at trough plasma concentrations) on day 3 of week 1. BTK occupancy was measured using an enzyme-linked immunosorbent assay. Free BTK proteins in the cell lysates were captured by a biotinylated-zanubrutinib probe coupled to plates coated with NeutrAvidin. Total BTK protein levels were measured in a parallel assay using plates coated with anti-BTK antibody (BD Biosciences) and detected by another anti-BTK antibody (Abcam). As this was a phase 1 (first-in-human) study of zanubrutinib, and given that quantitation of BTK occupancy was exploratory, no specific acceptance criteria were prespecified in the study protocol.
Clinical outcomes
End points for part 1 included a preliminary assessment of safety (including determination of the maximum tolerated dose), pharmacokinetics, and BTK occupancy in PBMCs. Part 2 end points included additional pharmacokinetic assessments, BTK occupancy in PBMCs and safety (all cohorts), an assessment of BTK occupancy in nodal tissues (cohort 2a and optionally for other part 2 patients), and a preliminary evaluation of efficacy in specific B-cell malignancies (including cohort 2a). Treatment-emergent adverse events (AEs) were collected until 28 days after the last dose of study treatment or until resolution of drug-related events, graded according to National Cancer Institute Common Terminology Criteria for Adverse Events, version 4.03, and coded using the Medical Dictionary for Regulatory Activities, version 19.0. Efficacy end points included overall response rate (ORR) and complete response (CR) and partial response (PR) rates, including PR with lymphocytosis (PR-L), using published criteria, including computed tomography imaging,20-22 as well as progression-free survival and overall survival. Bone marrow examination was required at screening and to confirm CR. Progression-free survival was defined as the duration from treatment initiation until disease progression or death; overall survival was defined as the time from treatment initiation until death. For the CLL/SLL cohorts, patients were considered evaluable for response if they were enrolled at least 3 months before the data cutoff date or if they had progressed before that date. All results are presented as of 15 September 2017, for dose-finding analysis, and 3 November 2017, for CLL/SLL safety and efficacy analyses.
Statistical analysis
The Kaplan-Meier method was used for time-to-event analyses with censoring. For response rates, 95% exact (Clopper-Pearson) confidence intervals (CIs) were provided. Toxicity data for all patients who received ≥1 dose of zanubrutinib were summarized using standard descriptive statistics for part 1/cohort 2a, and CLL/SLL patients separately.
Results
This first-in-human study of zanubrutinib in B-cell malignancies includes results from 144 patients, including 17 enrolled in part 1, 39 enrolled in cohort 2a, and 94 with CLL/SLL in part 2 (including 6 CLL/SLL patients from cohort 2a who were also included in dose-finding analyses; supplemental Figure 1). Independent audits for 1 of the trial sites revealed violations of good clinical practice involving 12 patients, including 8 enrolled in part 1. Consequently, all data from these 12 patients were excluded from the primary analyses reported here, and are summarized separately in supplemental Tables 2-5.
Dose finding
Patients enrolled in dose escalation (part 1) and cohort 2a comprised the dose-finding set (n = 56). Patient characteristics are shown in Table 1. In part 1, sequential patient cohorts received oral zanubrutinib at 40 mg once daily (n = 3), 80 mg once daily (n = 4), 160 mg once daily (n = 5), 320 mg once daily (n = 1), or 160 mg twice daily (n = 4). No dose-limiting toxicities were observed. Based on part 1 safety as well as pharmacokinetic and pharmacodynamic data, the maximally administered dose (either 320 mg once daily or 160 mg twice daily) was chosen for further study in part 2.
Demographics and baseline disease characteristics for all patients in part 1, cohort 2a, and patients with CLL/SLL in part 2
| Parameter | Dose escalation* | CLL/SLL† |
|---|---|---|
| Part 1: n = 17; and cohort 2a, part 2: n = 39 | Part 2: n = 94 | |
| Age, median (range), y | 67 (41-85) | 69 (24-87) |
| Sex | ||
| Male | 42 (75.0) | 73 (77.7) |
| Female | 14 (25.0) | 21 (22.3) |
| Race | ||
| White | 45 (80.4) | 86 (91.5) |
| Black or African American | 0 | 1 (1.1) |
| Asian | 9 (16.1) | 4 (4.3) |
| Other | 2 (3.6) | 3 (3.2) |
| ECOG performance status | ||
| 0 | 27 (48.2) | 47 (50.0) |
| 1 | 24 (42.9) | 42 (44.7) |
| 2 | 5 (8.9) | 5 (5.3) |
| Prior treatment status | ||
| Treatment-naive | 1 (1.8) | 22 (23.4) |
| Relapsed or refractory | 55 (98.2) | 72 (76.6) |
| No. of prior therapies, median (range)‡ | 2 (0-7) | 2 (1-9)§ |
| Cytogenetics, n/N evaluable|| | ||
| del(17p) or TP53 mutation | — | 18/94 (19.1) |
| del(11q) | — | 17/73 (23.3) |
| Unmutated IgHV | — | 14/21 (66.7) |
| Bulky disease, >10 cm | 0 | 5 (5.3) |
| Parameter | Dose escalation* | CLL/SLL† |
|---|---|---|
| Part 1: n = 17; and cohort 2a, part 2: n = 39 | Part 2: n = 94 | |
| Age, median (range), y | 67 (41-85) | 69 (24-87) |
| Sex | ||
| Male | 42 (75.0) | 73 (77.7) |
| Female | 14 (25.0) | 21 (22.3) |
| Race | ||
| White | 45 (80.4) | 86 (91.5) |
| Black or African American | 0 | 1 (1.1) |
| Asian | 9 (16.1) | 4 (4.3) |
| Other | 2 (3.6) | 3 (3.2) |
| ECOG performance status | ||
| 0 | 27 (48.2) | 47 (50.0) |
| 1 | 24 (42.9) | 42 (44.7) |
| 2 | 5 (8.9) | 5 (5.3) |
| Prior treatment status | ||
| Treatment-naive | 1 (1.8) | 22 (23.4) |
| Relapsed or refractory | 55 (98.2) | 72 (76.6) |
| No. of prior therapies, median (range)‡ | 2 (0-7) | 2 (1-9)§ |
| Cytogenetics, n/N evaluable|| | ||
| del(17p) or TP53 mutation | — | 18/94 (19.1) |
| del(11q) | — | 17/73 (23.3) |
| Unmutated IgHV | — | 14/21 (66.7) |
| Bulky disease, >10 cm | 0 | 5 (5.3) |
Values shown are n (%) unless otherwise indicated.
—, data not available; ECOG, Eastern Cooperative Oncology Group; IgHV, immunoglobulin variable-region heavy chain.
Includes the following B-cell malignancy histologies: CLL/SLL (n = 4), WM (n = 4), MCL (n = 6), diffuse large B-cell lymphoma (n = 2), and hairy cell leukemia (n = 1).
Includes 6 patients also included in cohort 2a.
Prior therapies included purine nucleoside analogs (fludarabine and pentostatin), alkylating agents (cyclophosphamide and chlorambucil), anti-CD20 antibodies (rituximab, ofatumumab, and obinutuzumab) and bendamustine either alone or in combination; anthracycline-based regimens (eg, cyclophosphamide, doxorubicin, vincristine, prednisone [CHOP]) and/or cyclophosphamide, vincristine, and prednisone [CVP]; alemtuzumab; and lenalidomide. Three patients had prior exposure to a BCL-2 inhibitor (venetoclax or navitoclax) and 2 patients received prior therapy with a phosphoinositide 3-kinase inhibitor (idelalisib or duvelisib).
Relapsed or refractory patients only.
Denominators are the number of patients with valid assessments.
Additional information regarding dose and schedule was derived from 39 patients enrolled in cohort 2a, a cohort designed specifically to assess BTK occupancy by zanubrutinib in lymph node tissue. In this cohort, patients were assigned to receive either 320 mg once daily or 160 mg twice daily. Patients were evaluated for safety and pharmacokinetic and pharmacodynamic properties in combination with those from part 1 to further define the optimal dosing schedule.
The most common AEs in the dose-finding set were upper respiratory tract infection (n = 22; 39.3%), contusion (n = 20; 35.7%), and cough and diarrhea (n = 15; 26.8% each). Grade ≥3 AEs reported in >2 patients were anemia (n = 7), pneumonia and pyrexia (n = 4 each), and acute kidney injury and neutropenia (n = 3 each; supplemental Table 6). At the cutoff date, 32 patients (57.1%) in the dose-finding set had discontinued study treatment, including 20 because of disease progression and 8 because of AEs (supplemental Table 7). Although numbers were small, no significant differences in AE profiles were observed between the 320 mg daily and 160 mg twice daily treatment schedules (supplemental Table 8).
Pharmacokinetics
Zanubrutinib is rapidly absorbed after oral administration with Cmax observed ∼2 hours after dosing (Figure 1). The Cmax and area under the concentration-time curve increased in a nearly dose-proportional manner from 40 to 320 mg after initial dose (supplemental Table 9). Mean Cmax was 346 and 658 ng/mL after a single dose of 160 and 320 mg, respectively. The mean half-life of zanubrutinib administered either as 160 mg twice daily or 320 mg once daily was ∼4 hours with minimal accumulation observed after repeated dosing.

Plasma concentration-time profile of zanubrutinib after initial doses of 40, 80, 160, and 320 mg once daily and 160 mg twice daily in part 1 and cohort 2a patients. Lines represent mean plasma concentration and error bars indicate standard deviation. *For the 160-mg twice-daily dose group, patients received only the morning dose (one-half of the daily dose) for the 24-hour pharmacokinetic evaluation on day 1 of week 1. BID, twice daily; QD, once daily.
Plasma concentration-time profile of zanubrutinib after initial doses of 40, 80, 160, and 320 mg once daily and 160 mg twice daily in part 1 and cohort 2a patients. Lines represent mean plasma concentration and error bars indicate standard deviation. *For the 160-mg twice-daily dose group, patients received only the morning dose (one-half of the daily dose) for the 24-hour pharmacokinetic evaluation on day 1 of week 1. BID, twice daily; QD, once daily.
Pharmacodynamics
BTK occupancy was assessed in PBMCs from 45 patients across all cohorts and in lymph nodes from 30 patients with available and adequate paired biopsy specimens. In PBMCs, complete or near complete (>95%) BTK occupancy was achieved starting at doses of 40 mg per day; BTK occupancy >95% was observed 4 hours postdose in almost all patients across all doses (Figure 2A). No significant differences were observed across dose groups. These results indicate that BTK remains fully occupied by zanubrutinib well after achieving peak plasma levels. Median (range) BTK occupancy on day 3 of week 1 (predose) in the 30 pairs of nodal tissue was 94% (82.4%-100%) in the 320-mg once-daily group (n = 12) and 100% (86.3%-100%) in the 160-mg twice-daily group (n = 18; P=.0189 via Mann-Whitney exact test). Nodal BTK occupancy was >95% in 50% of patients receiving 320 mg once daily and in 89% of patients receiving 160 mg twice daily (P =.0342 via Fisher exact test; Figure 2B). Based on the higher proportion of patients who achieved >95% sustained BTK occupancy in lymph nodes at trough concentrations of zanubrutinib, the 160-mg twice-daily regimen was determined to be the recommended phase 2 dose (RP2D).
![Figure 2. BTK occupancy. In (A) PBMCs and (B) nodal tissue. Data for individual patients at each time point are shown. Patient numbers and the percentage of patients with >95% BTK occupancy are noted. Triangles indicate percentages of BTK occupancy in PBMCs of patients predose, at 4 and 24 hours after zanubrutinib treatment on day 1 of week 1 (W1D1), predose on day 3 of week 1 (W1D3), and day 1 of week 2 (W2D1). BTK occupancy in lymph node biopsy specimens was assessed at baseline and predose on W1D3 and calculated as 1 − ([free BTK on day 3 predose] × [total BTK at screening])/([free BTK at screening] × [total BTK on day 3 predose]). Median values are shown as lines through the individual triangles. DLBCL, diffuse large B-cell lymphoma; FL, follicular lymphoma; MZL, marginal zone lymphoma.](https://ash.silverchair-cdn.com/ash/content_public/journal/blood/134/11/10.1182_blood.2019001160/7/m_bloodbld2019001160f2.png?Expires=1769089816&Signature=RsG610tpreOzFoD8HT4DXPhi61D45SMt6kTQa1RYz3HKcKMwvkTP9NILLTd5jijdyTR3xItXscHThsl0~7szmkS5tIFtCQtB6uYAjQzQlVFfUxKBu19FHkGrQZ6XE47Djgm9RlXB6brTvvpqWNQPGnefN30EB-18RbRiz8oYfRLQC4FFsnqLYSfxZJsxIuBa-5ZsCmiSX5P~bYaMFAsI0~mko3QHm5DLzpZkrOrfYJU2-L8eusOhbIm~Nmopm30RUMVeKtz8En8DIjOZXlUwha0UOxIj~EhQCqAWEaMP5icUjPUG54gXdNbTZcSQ7ElqDQm3ibK2ZZJsOM6LG-1S4A__&Key-Pair-Id=APKAIE5G5CRDK6RD3PGA)
BTK occupancy. In (A) PBMCs and (B) nodal tissue. Data for individual patients at each time point are shown. Patient numbers and the percentage of patients with >95% BTK occupancy are noted. Triangles indicate percentages of BTK occupancy in PBMCs of patients predose, at 4 and 24 hours after zanubrutinib treatment on day 1 of week 1 (W1D1), predose on day 3 of week 1 (W1D3), and day 1 of week 2 (W2D1). BTK occupancy in lymph node biopsy specimens was assessed at baseline and predose on W1D3 and calculated as 1 − ([free BTK on day 3 predose] × [total BTK at screening])/([free BTK at screening] × [total BTK on day 3 predose]). Median values are shown as lines through the individual triangles. DLBCL, diffuse large B-cell lymphoma; FL, follicular lymphoma; MZL, marginal zone lymphoma.
BTK occupancy. In (A) PBMCs and (B) nodal tissue. Data for individual patients at each time point are shown. Patient numbers and the percentage of patients with >95% BTK occupancy are noted. Triangles indicate percentages of BTK occupancy in PBMCs of patients predose, at 4 and 24 hours after zanubrutinib treatment on day 1 of week 1 (W1D1), predose on day 3 of week 1 (W1D3), and day 1 of week 2 (W2D1). BTK occupancy in lymph node biopsy specimens was assessed at baseline and predose on W1D3 and calculated as 1 − ([free BTK on day 3 predose] × [total BTK at screening])/([free BTK at screening] × [total BTK on day 3 predose]). Median values are shown as lines through the individual triangles. DLBCL, diffuse large B-cell lymphoma; FL, follicular lymphoma; MZL, marginal zone lymphoma.
CLL/SLL cohort
Clinical safety and efficacy data in this report include patients with CLL/SLL (n = 94) enrolled and treated in part 2, including 6 CLL/SLL patients enrolled in cohort 2a (Table 1). Twenty-two patients (23.4%) were treatment-naive. The median number of prior therapies for previously treated patients was 2 (range, 1-9). Some patients had high-risk disease features, including adverse cytogenetics [del(11q), 23.3%; del(17p) and/or TP53 mutation, 19.1%]. Patients enrolled in the 320-mg once-daily group (n = 40) were given the option of changing their treatment regimen to 160 mg twice daily after determination of the RP2D and schedule (supplemental Table 10). With a median follow-up of 13.7 months (range, 0.4-30.5 months), 89 patients (94.7%) are continuing study treatment. Two patients (both with untransformed CLL) have discontinued treatment because of progressive disease (1 who previously attained a PR and 1 with a best response of stable disease), whereas 2 others discontinued because of AEs, and 1 for unspecified reasons.
Safety
Most AEs were of grade 1 or 2 severity (Table 2). Grade 3/4 AEs reported in >1 patient were neutropenia (n = 6), anemia (n = 2), pneumonia (n = 2), and hypertension (n = 2). One patient had febrile neutropenia (grade 3) and 1 patient had disseminated herpes zoster infection (grade 3). Atrial fibrillation (grade 2) occurred in 1 patient with a history of hypertension and hyperlipidemia. Only 1 patient with CLL/SLL (receiving concomitant aspirin) experienced major hemorrhage (grade 3 subcutaneous hemorrhage). Concomitant antiplatelet (aspirin, clopidogrel, or nonsteroidal anti-inflammatory drugs) and anticoagulant (unfractionated or low-molecular-weight heparin, direct thrombin inhibitors, or warfarin) medications were used by 16.0% and 8.5% of patients, respectively. The exposure-adjusted incidence rate for grade 3 petechiae/purpura/contusion was 0.086 per 100 person-months. There were no deaths in the CLL/SLL cohort.
AEs occurring in patients with CLL/SLL in part 2
| AE | CLL/SLL, n = 94 | |
|---|---|---|
| Any grade | Grade 3/4 | |
| Any event, n (%) | 89 (94.7) | 34 (36.2) |
| Contusion | 33 (35.1) | 0 |
| Upper respiratory tract infection | 31 (33.0) | 0 |
| Cough | 24 (25.5) | 0 |
| Diarrhea | 20 (21.3) | 0 |
| Fatigue | 18 (19.1) | 0 |
| Back pain | 14 (14.9) | 1 (1.1) |
| Hematuria | 14 (14.9) | 0 |
| Headache | 13 (13.8) | 0 |
| Nausea | 13 (13.8) | 1 (1.1) |
| Rash | 12 (12.8) | 0 |
| Arthralgia | 11 (11.7) | 0 |
| Muscle spasms | 11 (11.7) | 0 |
| Urinary tract infection | 10 (10.6) | 1 (1.1) |
| Petechiae | 8 (8.5) | 0 |
| Constipation | 7 (7.4) | 0 |
| Purpura | 7 (7.4) | 1 (1.1) |
| Neutropenia | 7 (7.4) | 6 (6.4) |
| Pneumonia | 7 (7.4) | 2 (2.1) |
| Sinusitis | 7 (7.4) | 0 |
| Limb injury | 6 (6.7) | 0 |
| Abdominal pain | 5 (5.3) | 0 |
| Basal cell carcinoma | 5 (5.3) | 0 |
| Dizziness | 5 (5.3) | 0 |
| Dry mouth | 5 (5.3) | 0 |
| Peripheral edema | 5 (5.3) | 0 |
| Postprocedural contusion | 5 (5.3) | 0 |
| Hypertension | 5 (5.3) | 2 (2.1) |
| Cellulitis | 5 (5.3) | 1 (1.1) |
| Nasopharyngitis | 5 (5.3) | 0 |
| Squamous cell carcinoma of the skin | 5 (5.3) | 1 (1.1) |
| Anemia | 3 (3.2) | 2 (2.1) |
| AE | CLL/SLL, n = 94 | |
|---|---|---|
| Any grade | Grade 3/4 | |
| Any event, n (%) | 89 (94.7) | 34 (36.2) |
| Contusion | 33 (35.1) | 0 |
| Upper respiratory tract infection | 31 (33.0) | 0 |
| Cough | 24 (25.5) | 0 |
| Diarrhea | 20 (21.3) | 0 |
| Fatigue | 18 (19.1) | 0 |
| Back pain | 14 (14.9) | 1 (1.1) |
| Hematuria | 14 (14.9) | 0 |
| Headache | 13 (13.8) | 0 |
| Nausea | 13 (13.8) | 1 (1.1) |
| Rash | 12 (12.8) | 0 |
| Arthralgia | 11 (11.7) | 0 |
| Muscle spasms | 11 (11.7) | 0 |
| Urinary tract infection | 10 (10.6) | 1 (1.1) |
| Petechiae | 8 (8.5) | 0 |
| Constipation | 7 (7.4) | 0 |
| Purpura | 7 (7.4) | 1 (1.1) |
| Neutropenia | 7 (7.4) | 6 (6.4) |
| Pneumonia | 7 (7.4) | 2 (2.1) |
| Sinusitis | 7 (7.4) | 0 |
| Limb injury | 6 (6.7) | 0 |
| Abdominal pain | 5 (5.3) | 0 |
| Basal cell carcinoma | 5 (5.3) | 0 |
| Dizziness | 5 (5.3) | 0 |
| Dry mouth | 5 (5.3) | 0 |
| Peripheral edema | 5 (5.3) | 0 |
| Postprocedural contusion | 5 (5.3) | 0 |
| Hypertension | 5 (5.3) | 2 (2.1) |
| Cellulitis | 5 (5.3) | 1 (1.1) |
| Nasopharyngitis | 5 (5.3) | 0 |
| Squamous cell carcinoma of the skin | 5 (5.3) | 1 (1.1) |
| Anemia | 3 (3.2) | 2 (2.1) |
All AEs occurring in ≥5% of patients and grade ≥3 AEs occurring in 2 or more patients are reported.
Efficacy
Of the 94 patients with CLL/SLL, 78 were evaluable for response by virtue of having been enrolled at least 3 months before the data cutoff date or having progressed before that date. At a median follow-up of 13.7 months (range, 0.4-30.5 months), the ORR was 96.2% (95% CI, 89.2-99.2), including 2 patients (2.6%) who achieved a CR, 63 (80.8%) with PR, and 10 (12.8%) PR-L (Table 3). Early redistribution lymphocytosis was observed with return to baseline within 12 weeks in most patients (Figure 3C). All efficacy-evaluable patients with del(17p) or TP53 mutation responded (n = 16; ORR = 100% [95% CI, 79.4-100]). Response rates were comparable in treatment-naive patients and those with relapsed/refractory disease (ORR, 100% and 94.6%; CR rates, 4.5% and 1.8%, respectively; Table 3). Likewise, reductions in lymph node disease burden observed in treatment-naive patients (Figure 3A) and patients with relapsed/refractory disease (Figure 3B) were similar. There has been no incidence of Richter transformation. Median progression-free survival has not been reached and 12-month estimated progression-free survival is 100%; 2 patients have progressed at 15.3 and 16.4 months.
Response rates in evaluable patients with CLL/SLL in part 2
| Best response, n (%) [95% CI] | Treatment-naive, n = 22 | Relapsed or refractory, n = 56 | Total, N = 78 |
|---|---|---|---|
| ORR | 22 (100) [84.56-100] | 53 (94.6) [85.13-98.88] | 75 (96.2) [89.17-99.2] |
| CR | 1 (4.5) | 1 (1.8) | 2 (2.6) |
| PR | 18 (81.8) | 45 (80.4) | 63 (80.8) |
| PR-L | 3 (13.6) | 7 (12.5) | 10 (12.8) |
| SD | 0 | 2 (3.6) | 2 (2.6) |
| PD | 0 | 0 | 0 |
| Missing/not evaluable | 0 | 1 (1.8) | 1 (1.3) |
| Best response, n (%) [95% CI] | Treatment-naive, n = 22 | Relapsed or refractory, n = 56 | Total, N = 78 |
|---|---|---|---|
| ORR | 22 (100) [84.56-100] | 53 (94.6) [85.13-98.88] | 75 (96.2) [89.17-99.2] |
| CR | 1 (4.5) | 1 (1.8) | 2 (2.6) |
| PR | 18 (81.8) | 45 (80.4) | 63 (80.8) |
| PR-L | 3 (13.6) | 7 (12.5) | 10 (12.8) |
| SD | 0 | 2 (3.6) | 2 (2.6) |
| PD | 0 | 0 | 0 |
| Missing/not evaluable | 0 | 1 (1.8) | 1 (1.3) |
Sixteen patients were not included because the first protocol-specified response assessment time point of 3 months from treatment start date had not been reached and progression had not been observed.
PD, progressive disease; SD, stable disease.
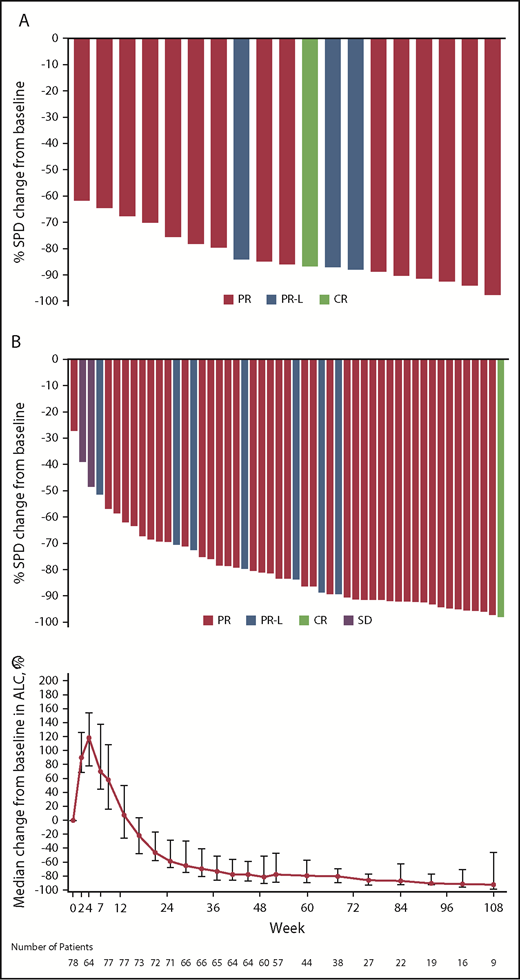
Reductions in the sum of the products of perpendicular diameters of target lymph nodes. In patients with (A) treatment-naive or (B) relapsed/refractory CLL/SLL; (C) kinetics of lymphocytosis. Data indicate median percentage change in lymphocyte counts and 95% CIs. ALC, absolute lymphocyte count; SD, stable disease; SPD, sum of the products of lymph node diameters as assessed by computed tomography scan.
Reductions in the sum of the products of perpendicular diameters of target lymph nodes. In patients with (A) treatment-naive or (B) relapsed/refractory CLL/SLL; (C) kinetics of lymphocytosis. Data indicate median percentage change in lymphocyte counts and 95% CIs. ALC, absolute lymphocyte count; SD, stable disease; SPD, sum of the products of lymph node diameters as assessed by computed tomography scan.
Discussion
Inhibition of BTK has proven remarkably effective as a mechanism-based therapeutic approach in B-cell malignancies, including CLL/SLL.23 Toxicities, some likely related to off-target effects, are the most frequent reason for treatment discontinuation in CLL patients treated with ibrutinib.7,11,24 The novel BTK inhibitor zanubrutinib has improved specificity for BTK compared with ibrutinib, with preclinical data showing improved selectivity (supplemental Table 1).13
Consistent with the favorable oral bioavailability in preclinical studies,13 oral administration of zanubrutinib achieved excellent plasma drug exposures with the RP2D of 160 mg twice daily. After adjusting for plasma protein binding (94.2% for zanubrutinib), this exposure is approximately eightfold higher than that observed with ibrutinib 560 mg daily.11,25 Furthermore, compared with acalabrutinib, another highly specific BTK inhibitor with demonstrated activity in CLL, zanubrutinib has a longer half-life (4 hours vs 1 hour, respectively),12 leading to more sustained exposure. This heightened exposure translates to complete inhibition of BTK in lymph node biopsy specimens and is hypothesized to maximize the chances of achieving deep and sustained remissions.
The constant BTK inhibition across blood and tissue compartments with zanubrutinib was associated with a favorable toxicity profile, in keeping with the drug’s high selectivity for BTK. Specifically, the relative sparing of EGFR from inhibition likely explains the lower rates of diarrhea and rash observed with zanubrutinib (∼20%, with no grade ≥3 AEs, compared with 42% to 51% for ibrutinib [Table 2]).5-7,9,10 Furthermore, although the mechanisms behind the major bleeding and atrial fibrillation associated with ibrutinib are not fully understood, these toxicities were infrequent, with each having been reported in only 1 patient in the current study. Lower-grade bleeding was still observed, most likely due to the on-target effect of BTK inhibition on platelet function, which is a class effect of all BTK inhibitors.16,26 However, zanubrutinib’s relative sparing of other kinases relevant to hemostasis, such as TEC, may favorably influence the incidence and severity of bleeding, consistent with our preliminary observations.27 In support of the hypothesis that highly specific BTK inhibition may lead to better drug tolerance, we note that only 2 patients in the CLL/SLL cohorts ceased study drug because of AEs. Additionally, lower rates of AEs have also been reported in patients treated with acalabrutinib.12
A unique aspect of this study is the detailed evaluation of the extent of BTK inhibition in lymph nodes. To our knowledge, a systematic evaluation of target inhibition in nodal tissues has not been undertaken with other BTK inhibitors. Notably, zanubrutinib achieves complete or near complete occupancy of BTK in lymph nodes at the RP2D. As BTK is irreversibly bound by all BTK inhibitors in clinical use, recovery of BTK function is solely a function of new protein synthesis. Therefore, it is not surprising that twice-daily dosing of zanubrutinib was more efficient in maintaining continuous BTK blockade in lymph nodes (100% median occupancy at trough plasma concentrations), and this led to the selection of 160 mg twice daily, rather than 320 mg once daily, as the RP2D on pharmacodynamic grounds. No clear differences in efficacy or safety were evident between the daily and split dose regimens. However, the power of the study to detect such differences was low given the high response rates with both regimens.
The clinical activity of zanubrutinib in CLL/SLL patients in this study was notable, with high ORRs in patients with treatment-naive (100%) and relapsed/refractory (93%) disease. Response rates with zanubrutinib were similarly high in patients regardless of high-risk cytogenetic features. CRs were uncommon at this early time point, consistent with previous experience with other BTK inhibitors.5,12,14,23,28 Longer follow-up is needed to more precisely determine the final complete remission rate and response duration for this CLL/SLL cohort. The fact that zanubrutinib and ibrutinib share the same binding site on BTK as ibrutinib suggests that cells bearing the C481S mutation may emerge with longer follow-up.
In this first-in-human study, zanubrutinib demonstrated encouraging activity in patients with relapsed/refractory and treatment-naive CLL/SLL, with good tolerability. Two ongoing randomized studies of zanubrutinib vs ibrutinib (NCT03053440 and NCT03734016) aim to determine whether consistent, continuous BTK blockade with a selective inhibitor results in fewer off-target effects and translates into improvements in disease control.
All authors had access to original data for the analyses described here. For original data, please contact: constantine.tam@petermac.org.
The online version of this article contains a data supplement.
The publication costs of this article were defrayed in part by page charge payment. Therefore, and solely to indicate this fact, this article is hereby marked “advertisement” in accordance with 18 USC section 1734.
Acknowledgments
The authors thank the patients who participated in the study, their supporters, and the investigators and clinical research staff from the study centers. Medical writing and editorial assistance were provided, under the direction of the authors, by Bio Connections.
This work, including medical writing and editorial assistance, was supported by BeiGene USA, Inc.
BeiGene was involved in study design, compilation of data, and statistical analysis.
Authorship
Contribution: Together with BeiGene authors (L.W., L.H., and J.H.), C.S.T., J.T., J.F.S., S.O., and A.W.R. were responsible for study design, and R.E., J.H., S.O., and C.S.T. contributed to data interpretation and analysis; all investigators (C.S.T., J.T., J.A.B., G.C., D.G., R.H., P.B.J., P.M., J.M., S.O., J.F.S., D.S., A.T., and A.W.R.) and their respective research teams reviewed patient records and contributed to data collection; BeiGene authors (R.E., Y.Y., Z.T., L.H., J.H., W.N., and L.W.) confirmed assay validation and data accuracy and compiled data for summation and analysis; Y.Y., Z.T., W.N., and L.W. performed data analysis and interpretation; C.S.T., A.W.R., R.E., and J.H. contributed to the first draft of the manuscript; C.S.T., R.E., and J.H. further contributed to final manuscript writing; C.S.T. had final responsibility to submit for publication; and all authors had full access to all of the data, carefully reviewed the manuscript, and approved the final version.
Conflict-of-interest disclosure: C.S.T. reports research funding, personal fees, and other from BeiGene during the conduct of the study, and personal fees from Janssen and Pharmacyclics outside of the submitted work. G.C. reports research funding from BeiGene during the conduct of the study, served on an advisory board for AbbVie Pty Ltd, and received travel grants from Amgen Australia and Takeda Australia outside of the submitted work. P.M. reports personal fees and nonfinancial support from Novartis, Roche, Janssen, Amgen, and Celgene outside of the submitted work. J.M. reports personal fees from Gilead/Kite Pharma, Pharmacyclics/Janssen, Bayer, Alexion, Pfizer, Juno/Celgene, Bristol-Myers Squibb, Genentech, and Kyowa outside of the submitted work. S.O. reports research funding from BeiGene during the conduct of the study. J.F.S. reports grants, personal fees, and other from AbbVie and Janssen, and personal fees and other from Acerta and Sunesis outside of the submitted work. D.S. reports grants from BeiGene during the conduct of the study; grants from Amgen, Acerta, Pharmacyclics, Millennium, Sanofi, and BeiGene; and grants and personal fees from AbbVie, Janssen, Roche, Celgene, and MSD outside of the submitted work. A.W.R. reports research funding from BeiGene during the conduct of the study; reports grants from AbbVie and Janssen; reports other from Genentech outside of the submitted work; and is an employee of the Walter and Eliza Hall Institute, which receives milestone and royalty payments relating to venetoclax. R.E., Z.T., L.H., J.H., and W.N. are employees of BeiGene USA. L.W. is an employee of BeiGene Beijing. Y.Y. is an employee of BeiGene Shanghai. The remaining authors declare no competing financial interests.
Correspondence: Constantine S. Tam, Peter MacCallum Cancer Centre, 305 Grattan St, Parkville, VIC 3050, Australia; e-mail: constantine.tam@petermac.org.

This feature is available to Subscribers Only
Sign In or Create an Account Close Modal