

In this issue of Blood, report the interaction between Down syndrome (DS), a major genetic leukemia predisposition condition, and inherited genetic alleles associated with increased susceptibility to childhood acute lymphoblastic leukemia (ALL).1
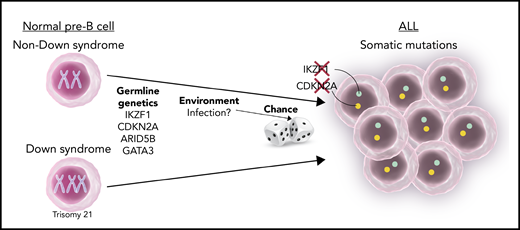
Multifactorial pathogenesis of childhood ALL. The development of childhood ALL is influenced by a combination of germline genetics, environmental factors (possibly associated with infection), and the chance occurrence of somatic mutations. DS is a major risk factor. The most significant allelic germline variants associated with ALL in children with and without DS are listed in the figure. Two of them (CDKN2A and IKZF1) are often also subjected to somatic loss-of-function mutations within the leukemic cells.
Multifactorial pathogenesis of childhood ALL. The development of childhood ALL is influenced by a combination of germline genetics, environmental factors (possibly associated with infection), and the chance occurrence of somatic mutations. DS is a major risk factor. The most significant allelic germline variants associated with ALL in children with and without DS are listed in the figure. Two of them (CDKN2A and IKZF1) are often also subjected to somatic loss-of-function mutations within the leukemic cells.
A recent study suggested that most cancers are caused by bad luck.2 They are initiated by somatic mutations occurring by chance in dividing stem cells during the natural process of DNA replication. Hence, the risk for specific cancers correlates with the accumulated number of cell divisions. This mechanism explains why ALL is the most common childhood cancer. The lymphoid system undergoes massive expansion during fetal development, in anticipation of exposure to infectious pathogens after birth. In addition, the process of V(D)J recombination, which enables antigen receptor diversity, is prone to generation of off-target genomic abnormalities. “Unlucky” mutations acquired during these developmental processes may initiate a preleukemic clone that is estimated to be present in at least 1 of every 20 newborns.3,4 However, the development of ALL requires the accumulation of additional somatic mutations, reducing the risk for pediatric ALL to ∼1 of every 2000 children.
This bad luck may be further aggravated by genetics. Genome-wide association studies (GWASs) have identified several, relatively common, genetic alleles whose presence increases the risk of ALL.5 Remarkably, germline variants that predispose to ALL occur in genes which play an important role in lymphoid development. These genes are also often modified by somatic mutations in the leukemic cells (see figure). Although statistically very significant, the added individual risk is small. Thus, a child carrying one of these alleles may double the risk for ALL, from ∼1:2000 to 1:1000.
In contrast, there are rare genetic predisposition syndromes that substantially increase the risk to leukemia. Of these, DS is the most common.6 The risk of ALL in DS is 20 times higher than in children without DS. Individuals with DS develop mostly B-cell precursor ALL. The most common genetic subtype of ALL in DS is characterized by activation of JAK-STAT signaling, caused by overexpression of the CRLF2 receptor. This subtype of ALL, generally associated with a worse prognosis, is found in ∼50% of ALLs in DS, compared with 5% to 10% of children without DS (herein “sporadic ALL”). How trisomy 21 contributes to the development of ALL, in particular CRLF2/JAK-STAT–driven ALL, is currently unknown.
Why do “only” 1% to 2% of children with DS develop ALL? Are there additional genetic factors that influence the risk of ALL in a child with DS? Are such genes unique to DS or similar to children with sporadic ALL? To answer these questions, Brown et al performed a meta-analysis of 4 independent GWASs comparing 542 DS-ALL with 1192 DS controls. Their major finding is that the same inherited genetic alleles that influence the risk to sporadic ALL are relevant to DS-ALL, although the magnitude of the added risk may differ between children with or without DS (see the article by Brown et al for details).
ALL is characterized by increased proliferation and self-renewal of lymphoid progenitors. Somatic mutations within the leukemic cells often involve genes that regulate normal lymphoid differentiation and proliferation. For example, CDKN2A, encoding a cell-cycle “brake,” is often somatically deleted in ALL, in particular, in CRLF2-ALLs, the most common subtype of DS-ALL. Brown et al found that the presence of an allelic variant of CDKN2A increased the risk to DS-ALL by 3.6, more than any other risk allele. Similarly, IKZF1, the most significant germline variant associated with an increased risk to either DS or sporadic ALLs (risk ratio of 2), is also subjected to loss-of-function somatic mutations in ALL, particularly in CRLF2-ALL and in DS-ALL (see figure). Brown et al show that this specific germline variant (rs58923657) resides in an enhancer and results in a lower expression of IKZF1. Interestingly, germline damaging variants in the IKZF1 coding region cause immune deficiency and an ALL predisposition syndrome (reviewed in Gocho and Yang7 ). Thus, there is a gradual increase in the risk associated with IKZF1 mutations, from a common germline enhancer variant that doubles the risk, through more damaging germline variants in coding regions that cause a cancer predisposition syndrome, to complete elimination of the gene, present as a somatically acquired event only in the leukemic cells.
An allelic variant of GATA3 that has previously been associated with CRLF2-ALL8 was also identified in the current study as a risk factor for DS-ALL (relative risk of 1.7). Previous studies have shown that this specific variant increases the expression of GATA3. GATA3 plays a major role in T-cell development and is specifically essential for type 2 innate lymphoid cells, key regulators of host immune response to parasitic infections and allergic inflammation. Interestingly, these cells express high levels of CRLF2 and activated JAK-STAT signaling.9 Thus, there may be a causal relationship between an inherited allele variant that increases the expression of GATA3 and an increased specific risk for CRLF2 expressing ALL in children with and without DS.
Although biologically fascinating, the identification of children at higher risk of developing ALL is currently of no clinical consequence. Theoretically, it may be possible to identify such children. Examples could be children with a combination of these genetic variants or children with genomic evidence of a preleukemic clones. However, what could be done with such information? Prevention of childhood leukemia is therefore a major scientific and clinical challenge. Some progress has been recently made in mouse models through modulating the microbial microenvironment.10 The genomic tools for identification of at-risk patients are already here. All that remains is to learn to alleviate bad luck.
Conflict-of-interest disclosure: The authors declare no competing financial interests.

This feature is available to Subscribers Only
Sign In or Create an Account Close Modal