


Abstract
β-Thalassemia and sickle cell disease (SCD) are the most prevalent monogenic diseases. These disorders are caused by quantitative or qualitative defects in the production of adult hemoglobin. Gene therapy is a potential treatment option for patients lacking an allogenic compatible hematopoietic stem cell (HSC) donor. New-generation lentiviral vectors (LVs) carrying a β-globin-like gene have revolutionized this field by allowing effective HSC transduction, with no evidence of genotoxicity to date. Several clinical trials with different types of vector are underway worldwide; the initial results are encouraging with regard to the sustained production of therapeutic hemoglobin, improved biological parameters, a lower transfusion requirement, and better quality of life. Long-term follow-up studies will confirm the safety of LV-based gene therapy. The optimization of patient conditioning, HSC harvesting, and HSC transduction has further improved the therapeutic potential of this approach. Novel LV-based strategies for reactivating endogenous fetal hemoglobin (HbF) are also promising, because elevated HbF levels can reduce the severity of both β-thalassemia and SCD. Lastly, genome-editing approaches designed to correct the disease-causing mutation or reactivate HbF are currently under investigation. Here, we discuss the clinical outcomes of current LV-based gene addition trials and the promising advantages of novel alternative therapeutic strategies.
β-Hemoglobinopathies
Hemoglobinopathies are the most frequent inherited diseases worldwide; it is estimated that 5% of the world’s population carries a defective hemoglobin (Hb) trait.1 The most common hemoglobinopathies are β-thalassemia and sickle cell disease (SCD). Patients affected by β-thalassemia show low or absent production of adult β-globin chains; this leads to α-globin/β-globin chain imbalance, death of erythroid cells, hemolysis, and iron overload.2,3 SCD is characterized by the production of a mutant β-globin chain (βS) that is incorporated in an Hb tetramer (HbS) that has a propensity to polymerize. This polymerization causes red blood cell (RBC) sickling, hemolysis, vaso-occlusive crises (VOCs), and acute chest syndrome.4
The geographical distribution of the β-globin gene mutations causing β-hemoglobinopathies is mainly driven by 2 factors: the endemicity of malaria and population movements. In fact, carriers are protected against malaria caused by Plasmodium falciparum, and high frequencies of β-globin gene variants are observed in sub-Saharan Africa, some Mediterranean regions, the Middle East, and India.5,6 Demographic changes (ie, migration from endemic areas to nonendemic areas) have spread these pathologies worldwide.1,7 Hence, the development of novel, effective, and safe therapies for β-hemoglobinopathies is a true challenge for the scientific and medical communities.
Understanding globin gene regulation to develop therapeutic strategies for β-hemoglobinopathies
The genes encoding the human α-like- and β-like globin chains are located on chromosomes 16 and 11, respectively, and are arranged in the order of their expression during development. The α-globin cluster consists of 3 genes (ζ [HBZ], α2 [HBA1], and α1 [HBA2]), and the human β-globin cluster consists of 5 genes (ε [HBE1], Gγ [HBG2], Aγ [HBG1], δ [HBD], and β [HBB]). Fetal Hb (HbF) is composed of 2 α-globin chains and 2 γ-globin chains. Fetal-to-adult Hb switching occurs soon after birth, and adult Hb (HbA) is composed of 2 α-globin chains and 2 β-globin chains. Several genetic elements are essential for the regulation of globin gene expression, including promoters and locus control regions (LCRs) containing DNase-hypersensitive sites (HSs). The α-globin LCR contains 4 enhancers, referred to as multispecies conserved sequences R1 to R4. The β-globin LCR contains 5 HSs (HS1 to HS5); HS2, HS3, and HS4 are the regulatory regions most involved in the stimulation of β-like globin gene transcription.8 The β-globin LCR regulates the expression of the β-like genes via looping-mediated interactions with the β-like globin promoters.9
Elevated levels of HbF are associated with less severe symptoms of both β-thalassemia and SCD.10 In fact, elevated HbF levels compensate for the HbA deficiency in β-thalassemia, whereas γ-globin exerts a potent antisickling effect in SCD. This observation fueled interest in factors that could modify the expression of the γ-globin genes. The BCL11A and LRF transcriptional factors occupy the γ-globin promoters in adult erythroid cells and have a major role in the repression of γ-globin gene expression.11,,,-15 Interestingly, several mutations in the γ-globin promoters associated with hereditary persistence of fetal Hb (HPFH)16 affect the binding of BCL11A and LRF,13,14 thus leading to elevated γ-globin expression.
Lentiviral approaches for treating β-hemoglobinopathies
Notwithstanding the high success rate of allogeneic hematopoietic stem cell (HSC) transplantation (HSCT), the transplantation of genetically corrected, autologous HSCs may be a curative treatment option for patients who lack an HLA-matched sibling donor. This approach avoids the immunological risks and toxicity associated with allogeneic HSCT.17 Research on gene therapy for hemoglobinopathies started ∼30 years ago. The idea was to engineer integrating retroviruses by replacing their genes with the transgene of interest. Firstly, pioneering studies used γ-retroviral vectors (γ-RVs) derived from the Moloney leukemia virus (MLV) to successfully transfer genes into murine repopulating stem/progenitor cell; however, levels of β-globin expression were low and nontherapeutic, and variegation of gene expression was observed.18 The discovery of the β-globin LCR (guaranteeing high-level, erythroid-specific expression of the β-like globin genes19 ) was fundamental in improving the design and therapeutic potential of these gene therapy vectors. However, the introduction of the LCR elements and the β-globin gene into MLV-based vectors was associated with proviral rearrangements20,21 The introduction of HIV-derived lentiviral vectors (LVs) constituted a major breakthrough, thanks to the LVs’ ability to accommodate complex transcriptional units and efficiently transduce HSCs. Furthermore, the use of self-inactivating vectors (ie, lacking the enhancer/promoter region in the long terminal repeats [LTRs]) has eliminated the risk of transactivation of the adjacent cellular genes by the viral cis-regulatory elements. Importantly, hot spots of MLV-based γ-RVs are highly enriched in proto-oncogenes, whereas LVs have a safer integration profile.22
In the early 2000s, 2 groups designed the first prototype LVs; TNS9 and HPV569 both contained large fragments of the human β-globin gene (including the native promoter) and LCR HSs. These vectors were able to correct disease features in transgenic mouse models of β-thalassemia23,24 and SCD,25 respectively (Figure 1). The HPV569 vector was used in the first human clinical trial approved worldwide for the gene therapy of transfusion-dependent β-thalassemia (TDT) and SCD (LG001; Table 1). This vector encodes a β-globin transgene (β -globinT87Q) harboring an amino acid derived from the γ-globin chain, which confers it with antisickling properties. To prevent the activation of adjacent genes by the LCR, 2 copies of the core of the chicken β-globin HS4 chromatin insulator were inserted into the viral LTRs.26 Importantly, the use of erythroid-specific and stage-specific regulatory elements restricted the risk of transactivation of endogenous genes to the late erythroid precursors, which then rapidly become transcriptionally inactive and enucleate. The first TDT patient successfully transplanted with LV-transduced autologous HSCs was a compound heterozygote for the βE allele (giving rise to low levels of an unstable tetramer [HbE]27 ) and a β0 allele (resulting in the absence of β-globin expression). After myeloablative conditioning with busulfan, the patient received 3.9 × 106/kg of transduced BM CD34+ hematopoietic stem/progenitor cells (HSPCs). The vector copy number (VCN) in the drug product was 0.6, and the patient became transfusion independent 12 months later. The level of Hb containing the transgene (HbAT87Q) rose progressively to 3.7 g/dL 3 years after gene therapy, which corresponded to more than one-third of the total Hb.26 At this follow-up time point, the patient produced a similar proportion of endogenous HbE (2.9 g/dL) and HbF (3.3 g/dL), which along with the HbAT87Q provided sustained levels of total Hb (9.9 g/dL). The transduction levels have remained stable for 9 years (mean VCN in peripheral blood mononuclear cells, 0.1); the patient has maintained stable levels of Hb and requires only occasional transfusions (M.C., unpublished data).28

The structure of LVs used in clinical trials for β-hemoglobinopathies. The LV name is given in bold type. HS2, HS3, and HS4 are hypersensitive sites from the LCR. ΔLTR, self-inactivating LTR; β-p, β-globin promoter; E, β-globin gene enhancer; FB, FII/BEAD-A; RRE, Rev-responsive element; SA, splice acceptor; SD, splice donor.
The structure of LVs used in clinical trials for β-hemoglobinopathies. The LV name is given in bold type. HS2, HS3, and HS4 are hypersensitive sites from the LCR. ΔLTR, self-inactivating LTR; β-p, β-globin promoter; E, β-globin gene enhancer; FB, FII/BEAD-A; RRE, Rev-responsive element; SA, splice acceptor; SD, splice donor.
Gene therapy clinical trials for TDT and SCD patients
| Trial number | Phase | Sponsor | Site | Start date/ recruitment status | Number of patients | Vector and transgene (nuclease and DP name) | Cell source | Conditioning | DP administration | Last update (www.clinicaltrials.gov) | References |
|---|---|---|---|---|---|---|---|---|---|---|---|
| β-Thalassemia | |||||||||||
| LG001 | 1/2 | bluebird bio | France | September 2006/completed | 2* | HPV569 (βA-T87Q-globin) | G-CSF mPBCs or BM | Myeloablative (busulfan) | IV | NA | 26 |
| NCT01639690 | 1 | Memorial Sloan Kettering Cancer Center | United States | July 2012/active, not recruiting | 4 | TNS9.3.55 (βA-globin) | G-CSF mPBCs | Nonmyeloablative (busulfan 8 mg/kg) | IV | 6 June 2018 | 113 |
| NCT02151526 (HGB205) | 1/2 | bluebird bio | France | July 2013/active, not recruiting | 4 | BB305 (βA-T87Q-globin) | G-CSF + plerixafor mPBCs | Myeloablative (busulfan) | IV | 31 January 2019 | 28 |
| NCT01745120 (HGB204) | 1/2 | bluebird bio | United States, Australia, Thailand | August 2013/completed | 18 | BB305 (βA-T87Q-globin) | G-CSF + plerixafor mPBCs | Myeloablative (busulfan) | IV | 8 May 2019 | 28 |
| NCT02453477 | 1/2 | IRCCS San Raffaele | Italy | May 2015/active, not recruiting | 10 | GLOBE (βA-globin) | G-CSF + plerixafor mPBCs | Myeloablative (thiotepa + threosulfan) | IO | 4 May 2018 | 33 |
| NCT02906202 (HGB207) | 3 | bluebird bio | United States, France, Germany, Greece, Italy, Thailand, United Kingdom, | July 2016/recruiting | 23 (estimated) | BB305 (βA-T87Q-globin) | G-CSF + plerixafor mPBCs | Myeloablative (busulfan) | IV | 31 January 2019 | 31 |
| NCT02906202 (HGB212) | 3 | bluebird bio | United States, France, Germany, Greece, Italy, Thailand, United Kingdom | June 2017/recruiting | 15 (estimated) | BB305 (βA-T87Q-globin) | G-CSF + plerixafor mPBC | Myeloablative (busulfan) | IV | 31 January 2019 | 31 |
| NCT03432364 | 1/2 | Sangamo Therapeutics and Bioverativ Therapeutics | United States | February 2018/recruiting | 6 | ZFN (ST-400) | mPBCs | Myeloablative (busulfan) | IV | 4 February 2019 | NA |
| NCT03655678 | 1/2 | Vertex Pharmaceuticals and CRISPR Therapeutics | Germany, United Kingdom | September 2018/recruiting | 12 (estimated; may be expanded to 45) | CRISPR/Cas9 (CTX001) | CD34+ human HSPCs (mobilization: NA) | Myeloablative (busulfan) | IV | 3 May 2019 | NA |
| SCD | |||||||||||
| NCT02151526 (HGB205) | 1/2 | bluebird bio | France | July 2013/active, not recruiting | 3 | BB305 (βA-T87Q-globin) | BM | Myeloablative (busulfan) | IV | 31 January 2019 | 37 |
| NCT02186418 | 1/2 | Children’s Hospital Medical Center, Cincinnati | United States, Jamaica | July 2014/recruiting | 10 | sGbG (γ-globin) | BM and plerixafor mPBCs | Reduced intensity conditioning (melphalan 140 mg/m2 BSA) | IV | 6 May 2019 | 44 |
| NCT02247843 | 1 | University of California Children’s Hospital, Los Angeles | United States | July 2014/recruiting | 6 | βAS3-FB (βAS3-globin) | BM | Myeloablative (busulfan) | IV | 29 March 2019 | NA |
| NCT02140554 (HGB206) | 1 | bluebird bio | United States | August 2014/recruiting | 50 (estimated; 3 groups [A, B, C]) | BB305 (βA-T87Q-globin) | BM (A and B) plerixafor mPBCs (C) | Myeloablative (busulfan) | IV | 20 May 2019 | 43 |
| NCT03282656 | 1 | David Williams, Boston Children’s Hospital | United States | February 2018/recruiting | 7 | BCH_BB-LCR shRNA(miR) shRNAmiR | Plerixafor mPBCs | Myeloablative (busulfan) | IV | 24 May 2018 | 54 |
| NCT03745287 | 1/2 | Vertex Pharmaceuticals Incorporated and CRISPR Therapeutics | United States | November 2018/recruiting | 12 (estimated; may be expanded to 45) | CRISPR/Cas9 (CTX001) | NA | Myeloablative (busulfan) | IV | 3 May 2019 | NA |
| Trial number | Phase | Sponsor | Site | Start date/ recruitment status | Number of patients | Vector and transgene (nuclease and DP name) | Cell source | Conditioning | DP administration | Last update (www.clinicaltrials.gov) | References |
|---|---|---|---|---|---|---|---|---|---|---|---|
| β-Thalassemia | |||||||||||
| LG001 | 1/2 | bluebird bio | France | September 2006/completed | 2* | HPV569 (βA-T87Q-globin) | G-CSF mPBCs or BM | Myeloablative (busulfan) | IV | NA | 26 |
| NCT01639690 | 1 | Memorial Sloan Kettering Cancer Center | United States | July 2012/active, not recruiting | 4 | TNS9.3.55 (βA-globin) | G-CSF mPBCs | Nonmyeloablative (busulfan 8 mg/kg) | IV | 6 June 2018 | 113 |
| NCT02151526 (HGB205) | 1/2 | bluebird bio | France | July 2013/active, not recruiting | 4 | BB305 (βA-T87Q-globin) | G-CSF + plerixafor mPBCs | Myeloablative (busulfan) | IV | 31 January 2019 | 28 |
| NCT01745120 (HGB204) | 1/2 | bluebird bio | United States, Australia, Thailand | August 2013/completed | 18 | BB305 (βA-T87Q-globin) | G-CSF + plerixafor mPBCs | Myeloablative (busulfan) | IV | 8 May 2019 | 28 |
| NCT02453477 | 1/2 | IRCCS San Raffaele | Italy | May 2015/active, not recruiting | 10 | GLOBE (βA-globin) | G-CSF + plerixafor mPBCs | Myeloablative (thiotepa + threosulfan) | IO | 4 May 2018 | 33 |
| NCT02906202 (HGB207) | 3 | bluebird bio | United States, France, Germany, Greece, Italy, Thailand, United Kingdom, | July 2016/recruiting | 23 (estimated) | BB305 (βA-T87Q-globin) | G-CSF + plerixafor mPBCs | Myeloablative (busulfan) | IV | 31 January 2019 | 31 |
| NCT02906202 (HGB212) | 3 | bluebird bio | United States, France, Germany, Greece, Italy, Thailand, United Kingdom | June 2017/recruiting | 15 (estimated) | BB305 (βA-T87Q-globin) | G-CSF + plerixafor mPBC | Myeloablative (busulfan) | IV | 31 January 2019 | 31 |
| NCT03432364 | 1/2 | Sangamo Therapeutics and Bioverativ Therapeutics | United States | February 2018/recruiting | 6 | ZFN (ST-400) | mPBCs | Myeloablative (busulfan) | IV | 4 February 2019 | NA |
| NCT03655678 | 1/2 | Vertex Pharmaceuticals and CRISPR Therapeutics | Germany, United Kingdom | September 2018/recruiting | 12 (estimated; may be expanded to 45) | CRISPR/Cas9 (CTX001) | CD34+ human HSPCs (mobilization: NA) | Myeloablative (busulfan) | IV | 3 May 2019 | NA |
| SCD | |||||||||||
| NCT02151526 (HGB205) | 1/2 | bluebird bio | France | July 2013/active, not recruiting | 3 | BB305 (βA-T87Q-globin) | BM | Myeloablative (busulfan) | IV | 31 January 2019 | 37 |
| NCT02186418 | 1/2 | Children’s Hospital Medical Center, Cincinnati | United States, Jamaica | July 2014/recruiting | 10 | sGbG (γ-globin) | BM and plerixafor mPBCs | Reduced intensity conditioning (melphalan 140 mg/m2 BSA) | IV | 6 May 2019 | 44 |
| NCT02247843 | 1 | University of California Children’s Hospital, Los Angeles | United States | July 2014/recruiting | 6 | βAS3-FB (βAS3-globin) | BM | Myeloablative (busulfan) | IV | 29 March 2019 | NA |
| NCT02140554 (HGB206) | 1 | bluebird bio | United States | August 2014/recruiting | 50 (estimated; 3 groups [A, B, C]) | BB305 (βA-T87Q-globin) | BM (A and B) plerixafor mPBCs (C) | Myeloablative (busulfan) | IV | 20 May 2019 | 43 |
| NCT03282656 | 1 | David Williams, Boston Children’s Hospital | United States | February 2018/recruiting | 7 | BCH_BB-LCR shRNA(miR) shRNAmiR | Plerixafor mPBCs | Myeloablative (busulfan) | IV | 24 May 2018 | 54 |
| NCT03745287 | 1/2 | Vertex Pharmaceuticals Incorporated and CRISPR Therapeutics | United States | November 2018/recruiting | 12 (estimated; may be expanded to 45) | CRISPR/Cas9 (CTX001) | NA | Myeloablative (busulfan) | IV | 3 May 2019 | NA |
BM, bone marrow; BSA, body surface area; CRISPR, clustered regularly interspaced short palindromic repeat; DP, drug product; G-CSF, granulocyte-colony stimulating factor; IO, intraosseously; mPBC, mobilized peripheral blood cell; NA, not available; shRNA, short hairpin RNA; ZFN, zinc-finger nuclease.
P1 failed to engraft and received the backup cells.
After the LG001 clinical trial, the vector was further optimized (Figure 1); in the BB305 transfer vector, the 5′ LTR promoter was replaced by the cytomegalovirus promoter (in order to boost viral production), and the chicken β-globin HS4 was removed. Indeed, the presence of the insulator within the LTR was associated with a low vector titer, genetic instability, and transient clonal dominance due to the presence of a cryptic splicing site that resulted in abnormal splicing of the high mobility group AT-hook 2 (HMGA2) gene.26
Three phase 1/2 clinical trials with the BB305 vector were approved: HGB-205 in France (for both TDT and SCD); HGB-204 in the United States, Australia, and Thailand (for TDT); and HGB-206 in the United States (for SCD) (Table 1).
The efficacy and safety of the BB305 LV in TDT patients were recently described.28,29 Twenty-two patients included in the HGB-204 or HGB-205 trial received mobilized, transduced CD34+ cells. After treatment, RBC transfusions could be withdrawn in 11 of the 13 patients with a non-β0β0 genotype, while the 2 other patients respectively showed 82% and 27% reductions in transfusion volume. Four of the 9 patients with a β0/β0 genotype or severe β+ homozygous mutations (eg, IVS-1-110, which decreases the production of normal HbA by ∼90%) became transfusion independent; the remaining 5 β0/β0 patients showed a median (range) reduction in transfusion volume of 53% (8% to 78%). Several patients showed normal or nearly normal levels of total Hb, together with the correction of ineffective erythropoiesis when Hb exceeded 9.5 g/dL. One patient no longer required iron chelation therapy and phlebotomies. The median (range) VCN after 15 months of follow-up was 0.3 (0.1-0.9) in the HGB-204 trial and 2 (0.3-4.2) in the HGB-205 trial. The VCN in the blood was well correlated with the production of therapeutic Hb. Transfusion independence in 3 patients with a β0/β0 or severe IVS1-110 genotype was achieved through a higher VCN or the endogenous production of HbF or HbA. However, these 3 patients did not attain normal levels of total Hb, and so the ineffective erythropoiesis was not fully corrected.
The BB305 vector is also being tested in 2 phase 3 clinical trials for TDT: HGB-207 for non-β0β0 genotypes and HGB-212 for β0/β0 genotypes. The main difference with regard to the above-mentioned phase 1/2 trials is the manufacturing process; 2 small molecules have been added to improve the transduction efficiency30 (Table 1). Promising initial data on the 11 treated TDT patients with a non-β0/β0 genotype were recently presented; the VCNs were higher than in previous clinical trials, and the vector’s safety has been confirmed. Ten of the 11 patients included in the HGB-207 trial were no longer receiving RBC transfusions, as was also the case for 2 of the 3 HGB-212 patients.31
Two other phase 1/2 clinical trials in patients with TDT have started in the United States (with the TNS9.3.55 vector) and Italy (with the GLOBE vector32 ); both LVs express the wild-type human β-globin gene (Table 1; Figure 1). The US study differs from above-mentioned trials because it uses a low-intensity conditioning regimen rather than a fully myeloablative conditioning regimen. The Italian trial features myeloablative conditioning with thiotepa and treosulfan rather than with busulfan. In the US trial, the VCN in the drug product and in vivo was low (0.2-0.4 and 0.02-0.08, respectively), and only 1 of the 4 patients had a reduced transfusion requirement.30 In the Italian trial, the VCN in the drug product was higher (0.7-1.5), and the mean VCN in erythroid precursors after 12 months of follow-up was 0.58. Transfusion requirement was reduced by ∼30 to 80% in the 3 adult patients. Three of the 4 pediatric patients became transfusion independent,33 and the transfusion requirement was reduced by ∼20% in the fourth patient. This was the first clinical trial for TDT to use intrabone infusion instead of IV injection in order to avoid the loss of HSCs in nontarget organs and accelerate hematopoietic engraftment. Even though the intrabone infusion route improved the engraftment in immunodeficient mice, the lack of a direct comparison between the 2 different administration routes in gene therapy clinical studies prevents one from drawing definitive conclusions. It is important to note that the transduced cells’ level of engraftment was higher in pediatric patients than in adult patients.33 Similarly, younger age has been linked to better outcomes in allogenic HSCT34 ; this might be due (at least in part) to damage to the BM caused by prolonged iron exposure in adult patients. This exposure is known to alter the iron sensing and storage machinery in mesenchymal stem cells (MSCs). The consequence is an accumulation of free iron and reactive oxygen species in the cytoplasm, which in turn decreases the number of MSCs and impairs the ability of MSCs to sustain hematopoietic function.35
The encouraging outcomes in our first β-thalassemia patients prompted us to attempt to treat 3 SCD patients in the HGB-205 trial. Prior to gene therapy, the patients had acute chest syndrome and recurrent VOCs and were in an RBC exchange program (as recommended by the French guidelines for severe forms of SCD36 ) after hydroxyurea treatment had proven to be ineffective.
The first βS/βS patient (1204) was successfully treated in France37 and no longer required conventional treatments (ie, RBC exchange and painkillers) 3 months later. After RBC exchange transfusions (to suppress endogenous erythropoiesis and improve the quality of the harvested CD34+ cells) and myeloablation with busulfan, the patient received 5.6 × 106/kg BM CD34+ cells with a VCN of ∼1. Fifteen months after treatment, HbAT87Q accounted for ∼50% of the total β-like globin chains, and the VCN in vivo was ∼2. The patient no longer experiences VOCs, displays a safe polyclonal hematopoietic reconstitution profile, and has laboratory parameters that are stable and similar to those observed in heterozygous SCD carriers.37
Two other patients with SCD were treated as part of the HGB-205 trial (patient 1207 [βS/βS] and patient 1208 [βS/β0]). They received a smaller dose of cells (4.7 × 106/kg and 3 × 106/kg, respectively) with a VCN <1 (2 drug products for each patient, with a VCN of 0.7/1 for patient 1207 and 0.8/0.5 for patient 1208). The levels of transduction in the peripheral blood were stable but lower than those observed for patient 1204 (0.4). Only 1 of the 2 patients (1208) became exchange-transfusion independent and symptom-free. Eighteen months after gene therapy, the protective Hb (∼2.7g/dL HbAT87Q and 1.6 g/dL HbF) accounted for ∼40% of total Hb (10.3 g/dL). No clinical benefit was observed in the second patient (1207), with 1.4 g/dL of HbAT87Q and 1.1 g/dL of HbF measured 14 months after gene therapy (total Hb = 7.8 g/dL 2 months after the last transfusion). The 2 patients who benefited from the gene therapy (1204 and 1208) showed a clear reduction in hemolysis marker levels and HbS polymerization and decreases in RBC membrane deformability and surface adherence.38 Importantly, we observed an elevation of β-globinT87Q and a decrease in βS-chain in mature RBCs relative to reticulocytes in all 3 treated patients, suggesting the strong, positive selection of transgene-expressing cells. This was probably because corrected RBCs had a longer half-life than unmodified RBCs in vivo.38 It is noteworthy that the patients with a successful treatment outcome presented some genetic characteristics relative to the βS/βS patient (1207). In fact, patient 1204 harbors a single 3.7-kb α-globin gene deletion; this might be associated with a small cytoplasmic pool of α-globin and a lower HbS content per cell39 and thus might have contributed to lower levels of RBC sickling. Patient 1208 has a βS/β0 genotype, which probably corresponds to a lower HbS content per cell than in βS/βS patients and, importantly, a high level of endogenous HbF.
The low CD34+ cell content after 2 BM collections (required for all 3 patients) prompted us to test the feasibility of plerixafor mobilization in patients with severe SCD in a phase 1/2 clinical trial. Three patients underwent plerixafor mobilization without experiencing adverse events. Large numbers of CD34+ cells (containing a high proportion of HSCs) were rapidly mobilized.40 Other groups have also obtained successful results when mobilizing HSCs in SCD patients with plerixafor.41
The results obtained in our first study encouraged us to treat other SCD patients with the same vector in the HGB-206 trial. In an initial group of 7 patients, the first results were unsatisfactory because of a low dose of CD34+ BM cells (median dose, 2.1 × 106/kg) and a low median VCN (0.3-1.3). Two years after the treatment, the VCN in the peripheral blood was ∼0.1, and HbAT87Q levels were <1 g/dL. The HbAT87Q accounted for <10% of the total Hb. A second group of 2 patients received preharvest RBC transfusions (as in the HGB-205 trial) and a cell dose of 2.2 or 3.2 × 106/kg. The drug product manufacturing process and the conditioning were optimized. Both patients showed a higher VCN (1.4 and 5) relative to the first group of 7 patients and a higher percentage of transduced cells (46% to 95%) in the drug product, enabling a higher VCN in the periphery (0.6 and 2.5), higher levels of total and therapeutic Hb, and lower levels of hemolysis markers.42 Lastly, in a third group of 14 patients, the use of plerixafor to safely harvest a large number of SCD HSCs was a key improvement. The first 6 of these patients received a mean dose of 7.1 × 106/kg CD34+ cells with a high VCN (4), and the preliminary results are promising.43 It is noteworthy that 3 years after gene therapy, a serious adverse event (myelodysplasia syndrome) was reported for 1 patient from the first group. Further analyses of patient’s cells did not show any evidence of LV-mediated insertional oncogenesis, demonstrating that this event was not due to the LV gene therapy and was probably triggered by the conditioning regimen.42
In the United States, 2 other phase 1/2 protocols for SCD have been initiated (Table 1; Figure 1). Malik et al used an LV encoding a modified γ-globin transgene to treat 2 βS/β0 patients after reduced-intensity conditioning. The early results appear to be encouraging; the safety profile is excellent, and LV-derived HbF expression accounted for ∼20% of the total Hb (VCN, 0.2-0.4).44 The patient with the longest follow-up (1 year) showed a reduction in anemia and chronic pain and no acute VOCs. In the second clinical trial, the vector contained a β-globin gene with 3 antisickling mutations.45 However, no results have yet been reported.
Although the comparison of different gene addition clinical trials is often difficult (given the study-specific variables such as the transfusion regimen at baseline), the data as a whole strongly suggest that the optimized drug manufacturing process, patient conditioning, cell dose, and HSC source represent key requirements for obtaining clinical benefit in gene therapy for β-thalassemia and SCD.
Along with these gene addition approaches, other promising LV-based approaches have been designed to increase levels of endogenous HbF. Deng et al generated an LV encoding a fusion protein between a zinc-finger domain (which binds to the γ-globin promoters) and LDB1 (a looping factor involved in the LCR/β-like promoters interaction)46 (Figure 1). Upon treatment of primary erythroblasts obtained from SCD patient blood samples in vitro, forced looping between the LCR and the γ-globin promoters led to γ-globin reactivation (with HbF accounting for up to ∼50% of the total Hb) and correction of the SCD cell phenotype with a low VCN per cell.47,48 Recently, this approach was tested in vitro in nonhuman primate HSPCs. Upon differentiation toward the erythroid lineage, the progeny of genetically modified nonhuman primate HSPCs produced elevated levels of HbF.49
The BCL11A transcriptional repressor is another potential target for reactivating HbF expression. Given that BCL11A knockdown has a negative impact in HSCs and B cells,50,-52 Brendel et al developed an LV that expresses a microRNA-adapted shRNA targeting BCL11A under the control of erythroid-restricted β-globin promoter/LCR HS2 and HS3 elements32 (Figure 1). HbF upregulation was observed in vitro in human primary erythroblasts and in vivo in a SCD mouse model.50,53 Based on these results, a phase 1 clinical trial for SCD was initiated in May 2018 (Table 1). An optimized transduction protocol led to a VCN of 3 to 5 in the drug product. Three months after transplantation, a SCD patient treated with plerixafor-mobilized, transduced HSPCs showed good levels of HbF (accounting for 23% of total Hb), and 60% of the RBCs expressed HbF.54 Although promising, these results will have to be confirmed in long-term studies, in view of the known HbF upregulation that occurs after allogeneic HSCT in patients with SCD and β-thalassemia, even in the event of graft failure.55,-57
Genome-editing approaches for treating β-hemoglobinopathies
Genome editing with designer nucleases (ie, ZFNs), transcription activator-like effector nucleases, and the CRISPR-associated nuclease Cas9 system) generates DNA double-strand breaks (DSBs) at specific genomic loci. In the cell, DSBs can be repaired through homology-directed repair (HDR), nonhomologous end-joining (NHEJ), or microhomology-mediated end-joining (MMEJ). HDR is a high-fidelity repair mechanism that allows the site-specific integration of a homologous DNA donor template. It can be exploited to potentially correct disease-causing mutations. The error-prone nuclease-mediated NHEJ and MMEJ pathways have mainly been exploited to obtain permanent gene inactivation and the disruption of cis elements regulating gene expression through the generation of small insertions and deletions.
In the field of β-hemoglobinopathies, strategies designed to correct disease-causing mutations or achieve therapeutic levels of fetal γ-globin expression exploit endogenous β-like gene promoters and the LCR to recreate expression patterns that are more physiologic than those associated with current vector-mediated gene addition approaches58,59 (Table 2). Indeed, LVs cannot accommodate the entire LCR required for high-level expression of the β-like globin genes; on average, a VCN of 1 in vitro produces therapeutic Hb levels that account for ∼20% of the total Hb.45,60
Comparison of LV-mediated gene addition and genome-editing strategies for treating β-hemoglobinopathies
| LV-mediated gene addition strategies | HDR-based genome-editing strategies | NHEJ-based genome-editing strategies | |
|---|---|---|---|
| Therapeutic gene expression | Low/intermediate (depending on the VCN) | High | High |
| Efficiency in human HSCs | High | Low/intermediate | High |
| Safety | High/intermediate | Potentially high | Potentially high |
| Genotoxic risks | Oncogene transactivation, generation of aberrant transcripts, gene inactivation | Off-targets, large genomic rearrangements, β-globin gene knockout (SCD) | Off-targets, large genomic rearrangements |
| Costs | High (viral delivery) | High (viral delivery)/low (nonviral delivery) | Low (nonviral delivery) |
| LV-mediated gene addition strategies | HDR-based genome-editing strategies | NHEJ-based genome-editing strategies | |
|---|---|---|---|
| Therapeutic gene expression | Low/intermediate (depending on the VCN) | High | High |
| Efficiency in human HSCs | High | Low/intermediate | High |
| Safety | High/intermediate | Potentially high | Potentially high |
| Genotoxic risks | Oncogene transactivation, generation of aberrant transcripts, gene inactivation | Off-targets, large genomic rearrangements, β-globin gene knockout (SCD) | Off-targets, large genomic rearrangements |
| Costs | High (viral delivery) | High (viral delivery)/low (nonviral delivery) | Low (nonviral delivery) |
HDR-based gene correction approaches have been investigated for the reversion of point mutations (eg, the A>T mutation that causes SCD61,-63 ) (Figure 2). Indeed, HDR-mediated modification was achieved in ≤40% of the β-globin alleles in HSPCs.61,62,64 Initial xenotransplantation studies showed a drastic decrease in the HDR frequency in vivo,61,64,65 which indicated a major difference in genome editing efficiency between a population composed mostly of progenitors (ie, HSPCs) on one hand and repopulating HSCs on the other. It is noteworthy that a lower genetic modification rate in HSCs than in HSPCs has also been described for LVs.60 However, the optimization of reagents, delivery methods and culture conditions has substantially improved the efficiency of HDR-mediated gene correction in HSCs.63,65,,-68 The frequency of HDR-based targeted integration within the β-globin gene was as high as 9% in bona fide HSCs.69 However, failed HDR-mediated gene correction can lead to gene disruption by NHEJ (a DNA repair pathway that is more active in HSCs), generating a β-thalassemic phenotype instead of correcting the SCD mutation.70 To minimize this risk, higher efficiency of HDR-mediated gene correction should be achieved. Importantly, the inclusion of selectable markers in the HDR donor template increased the yield of gene-corrected HSCs,63 although cell selection might result in the recovery of a small number of HSCs. Interestingly, administration of triplex-forming peptide nucleic acids and donor DNA to adult or fetal β-thalassemic mice was associated with (1) HDR-mediated β-globin gene correction in ≤10% of HSCs, and (2) an amelioration of the β-thalassemic phenotype.71,72 However, this approach led to a gene correction rate of only 5% in human HSPCs.71
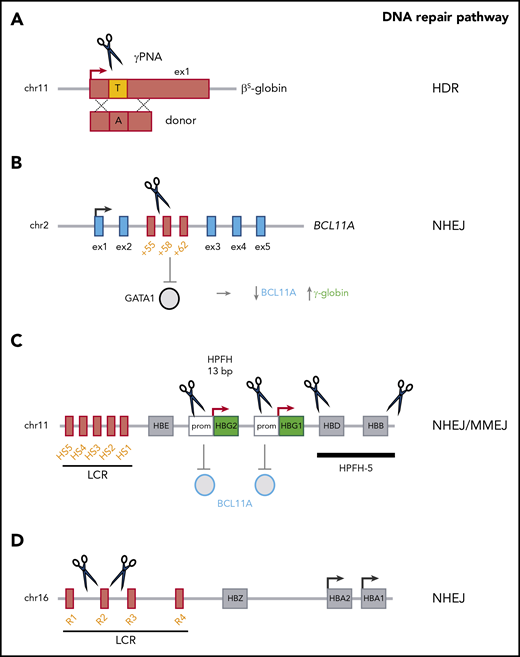
Genome-editing–based strategies for the treatment of β-hemoglobinopathies. (A) HDR-based approaches use nucleases or peptide nucleic acids (PNAs) and a donor template to correct the disease-causing mutation (eg, the SCD mutation). (B) BCL11A downregulation (via the NHEJ-mediated disruption of a GATA1 binding site in the +58-kb erythroid-specific enhancer) leads to γ-globin reactivation. (C) Mimicking HPFH mutations in the γ-globin promoter (Prom) (eg, the 13-bp deletion) through NHEJ and MMEJ leads to γ-globin reactivation, probably via BCL11A eviction. Reproducing large HPFH deletions via NHEJ (eg, HPFH-5) induces γ-globin expression. (D) Reducing α-globin expression (via the NHEJ-mediated deletion of the R2 α-globin enhancer) improves the α-/non-α-globin ratio in β-thalassemia. chr, chromosome; ex, exon.
Genome-editing–based strategies for the treatment of β-hemoglobinopathies. (A) HDR-based approaches use nucleases or peptide nucleic acids (PNAs) and a donor template to correct the disease-causing mutation (eg, the SCD mutation). (B) BCL11A downregulation (via the NHEJ-mediated disruption of a GATA1 binding site in the +58-kb erythroid-specific enhancer) leads to γ-globin reactivation. (C) Mimicking HPFH mutations in the γ-globin promoter (Prom) (eg, the 13-bp deletion) through NHEJ and MMEJ leads to γ-globin reactivation, probably via BCL11A eviction. Reproducing large HPFH deletions via NHEJ (eg, HPFH-5) induces γ-globin expression. (D) Reducing α-globin expression (via the NHEJ-mediated deletion of the R2 α-globin enhancer) improves the α-/non-α-globin ratio in β-thalassemia. chr, chromosome; ex, exon.
Lastly, NHEJ-based editing strategies, such as downregulation of the HbF repressor BCL11A73,74 or mimicking HPFH mutations,75,,-78 might be more efficient, given the apparent dominance of this pathway in HSCs.65,79 With a view to downregulating BCL11A gene expression in the erythroid lineage, 2 research groups have targeted the transcriptional activator GATA1’s binding site within an erythroid-specific BCL11A enhancer73,80,81 (Figure 2). In vitro, studies targeting the BCL11A erythroid enhancer using ZFNs or CRISPR/Cas9 have reported a high genome editing rate (up to ∼90%) and high γ-globin levels (accounting for up to ∼40% of total β-like globin chains) in erythroid cells derived from genetically modified adult HSPCs from healthy donors and patients with β-thalassemia or SCD.74,79,82 Elevated HbF levels were associated with an amelioration of the β-thalassemic and SCD cell phenotypes.79 Transplantation studies have shown that after ZFN or CRISPR/Cas9 transfection, BCL11A-edited HSPCs can stably engraft in immunodeficient mice, with an NHEJ frequency of ≤95%.74,79,82 These data are in line with the findings of other studies showing that NHEJ-mediated events observed in HSPCs in vitro are maintained in vivo in a high fraction of bona fide HSCs.65,69 It is noteworthy that complete inactivation of BCL11A impairs human RBC enucleation74 ; indeed, the results of recent xenotransplantation experiments by Chang et al suggest that BCL11A erythroid-enhancer editing is associated with a poor erythroid lineage reconstitution.83 However, individuals with BCL11A haploinsufficiency do not have defects in the erythroid lineage.84 Taken as a whole, these results indicate that safe genome-editing treatment approaches will require the fine modulation of BCL11A expression.
An alternative genome-editing approach to the treatment of β-thalassemia and SCD aims at generating HPFH mutations in HSCs and thus increasing HbF levels in the derived RBCs. In fact, HPFH is caused by 2 different types of mutations. Firstly, large genomic deletions encompassing the β- and δ-genes have been described in HPFH. Recreating deletional HPFH via NHEJ is feasible and effective75,77 but requires the generation of 2 DSBs, which might decrease the efficiency of genome editing (Figure 2). Secondly, HPFH mutations have been identified in the promoters10,14 of the 2 genes encoding the γ-globin chains (HBG1 and HBG2); they either generate de novo DNA motifs recognized by transcriptional activators85,-87 or disrupt binding sites for transcriptional repressors (eg, BCL11A13,14 ). A CRISPR/Cas9-based approach leading to the MMEJ-mediated generation of a naturally occurring HPFH 13-bp deletion spanning the BCL11A binding site was associated with significantly greater γ-globin expression (with HbF accounting for 35% of the total Hb) in primary-HSPC–derived RBCs from patients with SCD, together with correction of the SCD cell phenotype in vitro76 (Figure 2). However, given that the MMEJ and HDR repair pathways both use the same initial repair machinery,88 the MMEJ-based strategies might be less effective in bona fide HSCs.79 Targeting the BCL11A binding site in the γ-globin promoters has also been recently explored by other researchers.78,89 Importantly, loss of the HPFH 13-bp deletion was observed in vivo, although small deletions affecting the BCL11A-binding site (probably generated via NHEJ) and HbF reactivation were obtained in the progeny of bona fide HSCs.78,89 Notably, in this approach, the simultaneous cleavage of the 2 identical γ-globin promoters can result in the deletion of the intervening 5-kb genomic region and loss of the HBG2 expression (Figure 2). However, this deletion occurs at a low frequency in primary HSPCs.76,78
Lastly, an improvement in the typical α-/non-α-globin chain imbalance in β-thalassemia was recently achieved by the genome-editing–mediated downregulation of α-globin expression.90 The NHEJ-mediated CRISPR/Cas9 disruption of the multispecies conserved sequence R2 enhancer in the α-globin LCR led to downregulation of α-globin expression and decreased the α-/non-α-globin ratio in erythroid cells derived from β-thalassemic HSPCs (Figure 2). Moreover, this genomic modification was maintained in long-term repopulating HSCs.90
At present, there are no available data on the frequency of genome-edited HSCs in patients; although the preclinical data obtained in immunodeficient mouse models may have some predictive value, they may not always be translatable to patients. For example, the BM niche might be altered in patients.91 Clinical trials based on genome-editing strategies for β-hemoglobinopathies have been approved (Table 1). These trials’ results should provide insight into the potential benefits of these genome-editing therapeutic approaches relative to conventional gene addition by LVs.
Genotoxicity and gene therapy: challenges to be solved
In principal, genome-editing approaches are “targeted” because they are designed to modify a single, safe genomic target. Hence, they may be safer than an approach based on LVs that integrate semirandomly into the genome22 and may generate aberrant transcripts,26,92 which might potentially trigger oncogenesis.93 Although no genotoxic events have yet been observed in over 250 patients treated with LV-based gene therapy, confirmation of the safety of this approach will require a longer follow-up period. It is noteworthy that in a patient with chronic lymphocytic leukemia enrolled in a clinical trial using chimeric antigen receptor T cells, LV integration inactivated an oncosuppressor and led to clonal T cell expansion; fortuitously, the latter was beneficial for the patient’s clinical remission.94 Therefore, both the VCN and the type of integration must be carefully analyzed in order to monitor potential gene disruption or abnormal splicing events.
Although genome editing has a number of theoretical advantages over LV-mediated gene addition, potentially genotoxic nonspecific (“off-target”) DNA modifications might arise. However, high-fidelity Cas9 nucleases have been developed to minimize the frequency of off-target events.95,96 The generation of DSBs might also cause deletions, inversions, or translocations, even when the nuclease specifically recognizes a single target.97 Therefore, genome-wide off-target analyses98,99 and analyses of potential genomic rearrangements97,100 are mandatory for confirming the safety of genome-editing approaches in the treatment of β-hemoglobinopathies.
Importantly, HSCs are particularly sensitive to DSBs,101 especially in cases of multiple on-target events or concomitant on-target and off-target events. CRISPR/Cas9 treatment of HSPCs induces a DNA damage response that might contribute to the upregulation of apoptotic processes.102 However, a similar signature, along with delayed proliferation and lower HSC engraftment, is also elicited by LV transduction, although it is not dependent on DSBs caused by lentiviral integration.103 A CRISPR/Cas9 system targeting a single locus caused P53-dependent cell toxicity in induced pluripotent stem cells104 and cell-cycle arrest in immortalized retinal pigment epithelial cells; this resulted in the negative selection of cells with a functional P53 pathway.105 Therefore, P53 function should be carefully monitored in CRISPR/Cas9-modified cells used in gene therapy.
The genotoxicity and cytotoxicity potentially induced by designer nucleases could be avoided by using novel, targeted, DSB-free approaches.106,107 By way of an example, it was recently shown that a strategy based on the use of a Cas9-defective cytosine base editor was able to revert a β-thalassemic mutation in patient erythroblasts.108 Importantly, base editors have a lower level of off-target activity than Cas9 nuclease,109,110 and base editing occurs in quiescent cells, suggesting that bona fide HSCs could be genetically modified by applying this novel technology.111
In order to select the best therapeutic option, the efficacy and safety of genome-editing and LV gene addition strategies should be compared directly in preclinical studies. However, given the absence of reliable preclinical models of genotoxicity in long-term repopulating HSCs and their progeny, the safety of genome-editing approaches may ultimately have to be proven in clinical trials.
Conclusions
Given the disease and societal burden associated with β-hemoglobinopathies, there is an urgent need for effective, safe curative treatments. Gene therapy could potentially address this global health burden. Given the very high prevalence of β-hemoglobinopathies, no single therapeutic approach or product is likely to cover today’s unmet medical needs. Hence, the development of multiple, effective gene therapy approaches will be beneficial for society and the economy.
It is important to note that LVs are extremely expensive to manufacture (∼300 000 euros per patient112 ), and so the health care systems are unlikely to be able to pay for the treatment of large cohorts of patients. In most cases, genome-editing approaches require the delivery of RNA/protein reagents; they might be less expensive than LV-based strategies and thus would allow the broader use of gene therapy for these diseases. Moreover, the safety of genome-editing technologies (and base editing in particular) has improved dramatically; hence, one can reasonably suppose that editing approaches will replace LV-based gene therapy strategies in the coming years.
Acknowledgments
This project has received funding from the European Union’s Horizon 2020 research and innovation program under grant agreement 693762 (Gene For Cure). This work was also funded by the Agence Nationale de la Recherche (ANR-16-CE18-0004 and ANR-10-IAHU-01 “Investissements d’Avenir” program).
Authorship
Contribution: E.M., A.M., and M.C. wrote and edited the manuscript.
Conflict-of-interest disclosure: The authors declare no competing financial interests.
Correspondence: Marina Cavazzana, Assistance Publique-Hôpitaux de Paris, 149 rue de Sèvrese, 75015 Paris, France; e-mail: m.cavazzana@aphp.fr.
