

Key Points
Driver mutations in CALR or MPL encode for predicted neoantigens that bind MHC class I with high affinity in MPN patients.
The majority of MPN patients show evidence of recurrent candidate neoantigens, suggesting a potential use for targeted immunotherapy.

Abstract
Ph-negative myeloproliferative neoplasms (MPNs) are hematological cancers that can be subdivided into entities with distinct clinical features. Somatic mutations in JAK2, CALR, and MPL have been described as drivers of the disease, together with a variable landscape of nondriver mutations. Despite detailed knowledge of disease mechanisms, targeted therapies effective enough to eliminate MPN cells are still missing. In this study of 113 MPN patients, we aimed to comprehensively characterize the mutational landscape of the granulocyte transcriptome using RNA sequencing data and subsequently examine the applicability of immunotherapeutic strategies for MPN patients. Following implementation of customized workflows and data filtering, we identified a total of 13 (12/13 novel) gene fusions, 231 nonsynonymous single nucleotide variants, and 21 insertions and deletions in 106 of 113 patients. We found a high frequency of SF3B1-mutated primary myelofibrosis patients (14%) with distinct 3′ splicing patterns, many of these with a protein-altering potential. Finally, from all mutations detected, we generated a virtual peptide library and used NetMHC to predict 149 unique neoantigens in 62% of MPN patients. Peptides from CALR and MPL mutations provide a rich source of neoantigens as a result of their unique ability to bind many common MHC class I molecules. Finally, we propose that mutations derived from splicing defects present in SF3B1-mutated patients may offer an unexplored neoantigen repertoire in MPNs. We validated 35 predicted peptides to be strong MHC class I binders through direct binding of predicted peptides to MHC proteins in vitro. Our results may serve as a resource for personalized vaccine or adoptive cell–based therapy development.
Introduction
Ph-negative myeloproliferative neoplasms (MPNs) are clonal hematological malignancies with frequent mutations in JAK2, CALR, and MPL genes. Transformation to blast phase or secondary acute myeloid leukemia (post-MPN sAML) can occur. MPN patients testing negative for disease driver gene mutations are often referred to as “triple negative.”1,2 Targeted and whole-exome sequencing efforts have identified single nucleotide variants (SNVs) and small insertions and deletions (indels) with low overall mutation frequency in essential thrombocythemia (ET), polycythemia vera (PV), and primary myelofibrosis (PMF).1,3,4 Other mutation classes, such as gene fusions and splicing-related defects, have never been systematically mapped and explored in MPNs. RNA sequencing (RNA-seq) is the preferred method for the transcriptome-wide discovery of these mutation classes.5 One of the aims of our study was to extensively characterize the mutational landscape of MPN patients using a transcriptome-based approach.
Spontaneous T-cell responses of CD8+ and CD4+ T lymphocytes against MPN driver mutations JAK26 and CALR7,8 have been described, establishing MPNs as potentially immunogenic neoplasms and suggesting the use of personalized cancer vaccines, checkpoint inhibitors, adoptive T-cell therapy, or combinations thereof9 for the treatment of MPN. Neoantigens that also function as oncogenic driver mutations in the respective disease are highly attractive targets because they are shared between patients, essential for tumor survival, and expressed in the disease-causing clone.10-12 However, the huge diversity in HLA haplotypes significantly restricts the abundance of neoantigens. Therefore, further exploration of the neoantigen repertoire in MPN is essential to evaluate whether the MPN somatic mutational landscape offers the ability to identify candidate neoantigens for immunotherapy. In this study, we provide a framework for further systematic identification of MPN neoantigens for mutation classes, such as SNVs, indels, gene fusions, and splicing abnormalities,13 solely based on RNA-seq data obtained from granulocytes, the most abundant clonal myeloid cell type in MPN. Many of these mutation classes lead to frameshift mutations and the introduction of novel amino acid sequences; therefore, the distinction from the nonmutated protein sequence is more dramatic compared with SNVs. These mutation classes have also been shown to act as potent and specific immunogens.14,15 We predicted neoantigens for each patient in our MPN cohort on a personalized level, taking into account each patient’s MHC class I (MHCI) genotype. Finally, we validated the peptide:MHCI binding affinity for a subset of candidate neoantigenic peptides and, thereby, established a resource for immunotherapy in MPN.
Material and methods
Patient and control samples
A total of 104 MPN patients and 15 healthy controls was recruited into the study at the Medical University of Vienna, and 9 post-MPN sAML patients were recruited from the Fondazione IRCCS Policlinico San Matteo (Figure 1A; Table 1). The study was reviewed and approved by local ethics committees, and all patients and healthy controls provided informed consent in accordance with the Declaration of Helsinki. Criteria applied for the diagnosis of patients were described previously1 (supplemental Table 1, available on the Blood Web site).
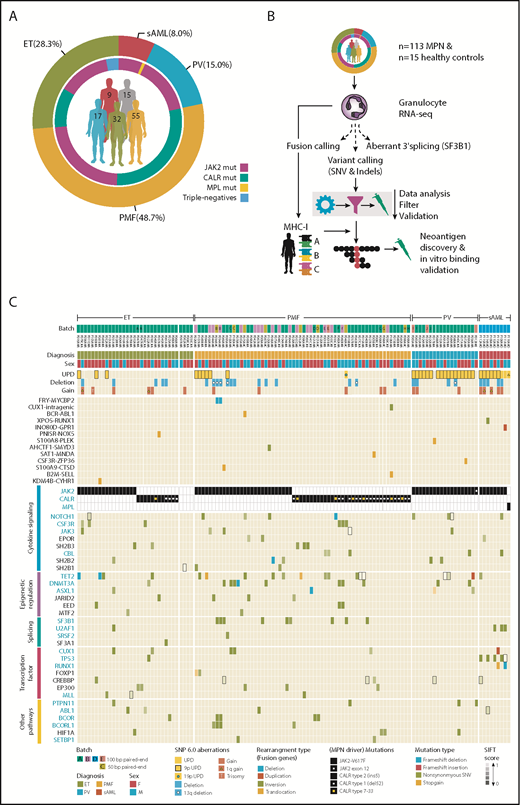
Large aberrations and small mutations (SNVs and indels) shape the MPN mutational landscape. (A) Distribution of diagnosis within the MPN cohort (outer ring). Occurrence of the MPN driver mutation status within each diagnosis (inner ring). Number of patients with each diagnosis (center). Healthy controls are in gray. (B) Transcriptome sequencing was performed on granulocyte RNA from 113 MPN patients and 15 healthy controls. Independent workflows for fusion calling, variant calling (SNVs and indels), and differential splicing analysis (in SF3B1-mutated patients) were established. Mutations were called, filtered, and validated; those leading to protein changes were translated to amino acid sequences. Neoantigens were predicted on a personalized level, taking each patient’s MHCI haplotype into consideration. (C) A total of 123 MPN patient samples is depicted and sorted by diagnosis, MPN driver mutation status (JAK2, CALR, or MPL positive), and nondriver mutation frequency. Patient sample replicates (n = 10) sequenced across or with the same batch (A-E) are indicated in capital letters (A-J). Chromosomal aberrations panel: uniparental disomies (UPDs), deletions, and gains were called with Affymetrix SNP 6.0 arrays (supplemental Table 5). Fusion panel: fusions were private among patients and colored by rearrangement type. Two fusions were reported for patient P106A#A. For fusion calling, we combined the results of 3 fusion detection tools (deFuse, SOAPfuse, and TopHat-Fusion) to overcome algorithm-specific biases, a practice that is frequently suggested in fusion benchmarking studies.39,40 MPN driver mutation panel: MPN driver mutation status (JAK2, CALR, and MPL) was determined as described in supplemental Methods. SNVs and indels panel: genes are grouped by occurrence in pathways and by mutation frequency. Mutations are colored by mutation type, and a gradient for high and low SIFT score was applied (only for SNVs). Small rectangles with black frame enhance visibility for mutations with low SIFT score. Only those genes with ≥2 mutations across the cohort, with the exception of genes involved in the splicing machinery (SRSF2, SF3A1), are shown. Gene names in blue are part of the TruSight Myeloid Sequencing Panel. For validation, variants from 77 RNA/DNA pairs were compared (supplemental Figure 6). Of 113 variants, 91 (81%) were concordant between RNA and DNA, 6 (5%) variants were only called on the RNA level, and 16 (14%) variants were only called on the DNA level (supplemental Table 10). Of the 16 variants called on DNA only, 4 were filtered out because of RNA-specific filters (supplemental Methods: RNA-specific filter a-e), and the remaining 12 were not called because of low gene expression and low variant allele frequency.
Large aberrations and small mutations (SNVs and indels) shape the MPN mutational landscape. (A) Distribution of diagnosis within the MPN cohort (outer ring). Occurrence of the MPN driver mutation status within each diagnosis (inner ring). Number of patients with each diagnosis (center). Healthy controls are in gray. (B) Transcriptome sequencing was performed on granulocyte RNA from 113 MPN patients and 15 healthy controls. Independent workflows for fusion calling, variant calling (SNVs and indels), and differential splicing analysis (in SF3B1-mutated patients) were established. Mutations were called, filtered, and validated; those leading to protein changes were translated to amino acid sequences. Neoantigens were predicted on a personalized level, taking each patient’s MHCI haplotype into consideration. (C) A total of 123 MPN patient samples is depicted and sorted by diagnosis, MPN driver mutation status (JAK2, CALR, or MPL positive), and nondriver mutation frequency. Patient sample replicates (n = 10) sequenced across or with the same batch (A-E) are indicated in capital letters (A-J). Chromosomal aberrations panel: uniparental disomies (UPDs), deletions, and gains were called with Affymetrix SNP 6.0 arrays (supplemental Table 5). Fusion panel: fusions were private among patients and colored by rearrangement type. Two fusions were reported for patient P106A#A. For fusion calling, we combined the results of 3 fusion detection tools (deFuse, SOAPfuse, and TopHat-Fusion) to overcome algorithm-specific biases, a practice that is frequently suggested in fusion benchmarking studies.39,40 MPN driver mutation panel: MPN driver mutation status (JAK2, CALR, and MPL) was determined as described in supplemental Methods. SNVs and indels panel: genes are grouped by occurrence in pathways and by mutation frequency. Mutations are colored by mutation type, and a gradient for high and low SIFT score was applied (only for SNVs). Small rectangles with black frame enhance visibility for mutations with low SIFT score. Only those genes with ≥2 mutations across the cohort, with the exception of genes involved in the splicing machinery (SRSF2, SF3A1), are shown. Gene names in blue are part of the TruSight Myeloid Sequencing Panel. For validation, variants from 77 RNA/DNA pairs were compared (supplemental Figure 6). Of 113 variants, 91 (81%) were concordant between RNA and DNA, 6 (5%) variants were only called on the RNA level, and 16 (14%) variants were only called on the DNA level (supplemental Table 10). Of the 16 variants called on DNA only, 4 were filtered out because of RNA-specific filters (supplemental Methods: RNA-specific filter a-e), and the remaining 12 were not called because of low gene expression and low variant allele frequency.
Description of the patient cohort and clinical parameters
| Controls | ET | PMF | PV | Post-MPN sAML | |
|---|---|---|---|---|---|
| WHO classification (2016) | |||||
| n (%) | 15 (11.7%) | 32 (25.0%) | 55 (43.0%) | 17 (13.3%) | 9 (7.0%) |
| Demographics | |||||
| Sex, n | 7 (f), 8 (m) | 21 (f), 11 (m) | 29 (f), 26 (m) | 9 (f), 8 (m) | 4 (f), 5 (m) |
| Age, mean ± SD, y | 37.4 ± 11.2 | 67.2 ± 11.8 | 65.5 ± 12.8 | 64.4 ± 11.0 | 74.3 ± 8.2 |
| Genotype | |||||
| Driver mutation status, n | NA | 17 JAK2-V617F 11 CALR (type 1 & 2) 4 triple-negative | 26 JAK2-V617F 29 CALR (type 1 & 2) | 16 JAK2-V617F 1 JAK2 exon 12 | 8 JAK2-V617F 1 MPL-W515A |
| Blood parameters at diagnosis | |||||
| Hemoglobin level, g/dL | NA | 14.0 ± 1.3 | 12.6 ± 1.8 | NA | NA |
| White blood cell count, g/dL | NA | 9.4 ± 2.8 | 9.9 ± 5.3 | NA | NA |
| Platelet count, ×109/L | NA | 853.6 ± 294.1 | 762.7 ± 444.6 | NA | NA |
| Outcome | |||||
| Overall survival, mean ± SD, mo | NA | 185 ± 77 | 158 ± 87 | 145 ± 86 | NA |
| Controls | ET | PMF | PV | Post-MPN sAML | |
|---|---|---|---|---|---|
| WHO classification (2016) | |||||
| n (%) | 15 (11.7%) | 32 (25.0%) | 55 (43.0%) | 17 (13.3%) | 9 (7.0%) |
| Demographics | |||||
| Sex, n | 7 (f), 8 (m) | 21 (f), 11 (m) | 29 (f), 26 (m) | 9 (f), 8 (m) | 4 (f), 5 (m) |
| Age, mean ± SD, y | 37.4 ± 11.2 | 67.2 ± 11.8 | 65.5 ± 12.8 | 64.4 ± 11.0 | 74.3 ± 8.2 |
| Genotype | |||||
| Driver mutation status, n | NA | 17 JAK2-V617F 11 CALR (type 1 & 2) 4 triple-negative | 26 JAK2-V617F 29 CALR (type 1 & 2) | 16 JAK2-V617F 1 JAK2 exon 12 | 8 JAK2-V617F 1 MPL-W515A |
| Blood parameters at diagnosis | |||||
| Hemoglobin level, g/dL | NA | 14.0 ± 1.3 | 12.6 ± 1.8 | NA | NA |
| White blood cell count, g/dL | NA | 9.4 ± 2.8 | 9.9 ± 5.3 | NA | NA |
| Platelet count, ×109/L | NA | 853.6 ± 294.1 | 762.7 ± 444.6 | NA | NA |
| Outcome | |||||
| Overall survival, mean ± SD, mo | NA | 185 ± 77 | 158 ± 87 | 145 ± 86 | NA |
WHO, World Health Organization.
PV patients were all JAK2 positive (16 JAK2-V617F, 1 JAK2 exon 12), 26 PMF patients were JAK2-V617F positive and 29 were CALR positive (17 type 1 or type 1-like, 12 type 2 or type 2-like), 17 ET patients carried JAK2 mutations and 11 carried CALR mutations (4 type 1, 7 type 2 or type 2-like), and 4 patients were triple-negative cases. All post-MPN sAML patients were JAK2-V617F positive, with the exception of 1 patient with an MPL (W515A) mutation (Figure 1A; supplemental Table 1).
Transcriptome library preparation and sequencing
Total RNA was isolated, applying standard procedures, from peripheral blood granulocytes. Next-generation sequencing of complementary DNA libraries of polyA-enriched RNA was performed using different library preparation kits and sequenced in various configurations on an Illumina HiSeq 2000 instrument, as listed in supplemental Table 2.
Driver mutation analysis
The presence of mutations and the mutational burden of JAK2, MPL, and CALR were determined in all patients, as described by Klampfl et al.1
Transcriptome data analysis for fusion detection and validation
Sequencing reads were mapped and processed independently with fusion detection tools deFuse, TopHat-Fusion, and SOAPfuse using human genome version GRCh37 or hg19. The results of these fusion-detection algorithms were merged and filtered further, as described in supplemental Methods. The fusion candidates that passed the filtering steps were validated by reverse-transcription polymerase chain reaction using RNA isolated from human peripheral granulocytes, followed by Sanger sequencing of the polymerase chain reaction products, as described in supplemental Methods.
Variant calling on RNA-seq data and targeted resequencing of patients’ genomic DNA
For the purpose of identification of SNVs and indels, we developed a workflow combining the Genome Analysis Toolkit’s (version 3.4-46)16 “Best Practices workflow for RNAseq” with in-house algorithms for reducing false-positive calls (see supplemental Methods). Variants were filtered to enrich for somatic mutations, as described in supplemental Methods. For 77 patients, additional targeted resequencing of genomic DNA was performed (supplemental Methods).
Estimating expression values on the gene level and differential expression analysis
Sequencing reads were trimmed (Trimmomatic) and aligned with the STAR aligner to the UCSC hg38 reference genome. Reads overlapping transcript features were counted with the summarizeOverlaps function of Bioconductor library GenomicAlignments. A detailed description of the workflow is described in supplemental Methods. The Bioconductor package DESeq2 was then used to model the data set and call differentially expressed genes.
Identification of aberrant 3′ splicing in SF3B1-mutated patients and validation
RNA-seq reads were aligned to the human genome, as described above in the section entitled “Variant calling on RNA sequencing data and targeted resequencing of patients’ genomic DNA.” Splice junction coverage files SJ.out.tab from STAR aligner were used for further splice junction analysis. Aberrant 3′ splicing was detected following published instructions.17 Novel splice junctions were defined as described in supplemental Methods. Differential splice site usage was tested with the testForDEU function of the DEXSeq Bioconductor package (version 1.14.2). Obtained P values were adjusted for false discovery rate using the Benjamini-Hochberg procedure. Hierarchical clustering was performed using the R package pheatmap. Percent-spliced-in (PSI) scores for alternative 3′ splice sites (3′ss) were calculated as described in Figure 4A. Aberrant 3′ss were validated with fragment length analysis using capillary electrophoresis, as described in supplemental Methods.
Neoepitope prediction and prioritization
We generated FASTA sequences of peptides from mutation classes, such as SNVs, indels, fusions, and aberrantly spiced genes, as described in supplemental Methods. Predicted HLA-I alleles for each patient (seq2HLA,18 release 2.2) and the mutated peptide sequences were used as input for NetMHCpan19 (version 4.0) to predict the binding affinities of 8-, 9-, and 10-mer peptides to MHCI proteins. We further filtered the NetMHCpan results based on criteria defined in supplemental Methods.
Peptide:MHCI binding validation through peptide-exchange technology
HLA Flex-T monomers (HLA*A03:01, HLA*A11:01, HLA*B07:02, HLA*B08:01), as well as positive and negative control peptides with known binding affinities, were purchased from BioLegend. The peptides were custom designed by Eurogentec. Monomers were mixed 1:1 with the appropriate peptides, incubated for 30 minutes under UV light (365 nm), and then kept in the dark for 30 minutes. A total of 1 µL of this mixture was used 1:1000 in duplicates in the enzyme-linked immunosorbent assay (ELISA) following the Flex-T HLA Class I ELISA protocol from BioLegend. The read-out is shown as mean absorbance.
Statistical analysis
Pairwise associations of genes and diseases were calculated using Fisher’s exact test followed by false discovery rate correction (Benjamini-Hochberg). All statistical analyses were performed using R (version 3.2.3 2015-12-10).
Results
Using whole-transcriptome sequencing, we analyzed RNA from granulocytes of 104 patients with chronic MPN (32 ET, 17 PV, 55 PMF) and 9 patients with disease that transformed to post-MPN sAML, as well as 15 healthy controls (n = 128). (Figure 1A; Table 1; supplemental Tables 1 and 2). We established a transcriptome-based bioinformatics workflow to enable the identification of gene fusions, variants (SNVs and indels), and splicing abnormalities (Figure 1B, supplemental Figure 1).
Fusion discovery
To establish the fusion discovery workflow, we first performed RNA-seq of 20 patient samples from various hematological malignancies with known fusion genes validated with fluorescence in situ hybridization. With the exception of fusion rearrangements involving promoter exchange (most IGH fusions), which are known to be difficult to capture with RNA-seq, we were able to recover all validated fusions and identify novel fusion partners (supplemental Figure 2; supplemental Table 3). Next, we applied the same workflow to the MPN patient cohort and identified a total of 13 fusions that passed all filtering criteria (supplemental Figure 3A-F; supplemental Table 4). All complementary DNA breakpoints were validated by Sanger sequencing (supplemental Figure 4). Eleven of 13 fusions were detected in chronic-phase MPN, and 2 were detected in post-MPN sAML patients (Figure 1C; supplemental Figure 5A). The majority (8/13) of fusions had fusion partners in trans (ie, on different chromosomes) and, therefore, were suspected to be caused by translocations. The remaining 5 fusions were formed in cis by inversions (n = 3), by a large-scale deletion (n = 1), and by a tandem duplication (n = 1) (supplemental Figure 5A-B).
Fusion discovery results were merged with cytogenetic aberrations data obtained from Affymetrix SNP 6.0 arrays to verify whether breakpoints coincided (supplemental Figure 5B; supplemental Tables 4 and 5). A large chromosome 13q deletion detected by single nucleotide polymorphism arrays in a PMF patient led to a FRY-MYCBP2 gene fusion (supplemental Figure 4D). None of the other fusion genes overlap with previously detected chromosomal aberrations or were not within the detection range of single nucleotide polymorphism arrays. RNA expression values for all fusion genes were low (mean, 3.2; standard deviation, 2.3; per kilobase of seed region and per million mapped reads) with the exception of FRY-MYCBP2 (106.7 per kilobase of seed region and per million mapped reads) (supplemental Figure 5C); however, the respective wild-type gene expression value for each gene-forming part of the fusion varied considerably (supplemental Figure 5D).
Among the 13 fusions found in MPN and post-MPN sAML patients, BCR-ABL1 t5,19 and INO80D-GPR1 inv2 were the only in-frame fusions (supplemental Figure 4A,H). For the INO80D-GPR1 inversion, we identified the genomic breakpoint region to be in intron 8 (genomic position chr2:206 584 716; hg19) of INO80D and exon 4 (chr2:206 750 070; hg19) of GPR1 (supplemental Figure 4I). We screened an additional 96 MPN patients for the presence of INO80D-GPR1; however, no other occurrence was found, suggesting that this inversion is likely a private somatic aberration.
Variant calling at transcriptome-level and mutation frequencies in MPN
Next, we applied the variant discovery workflow to RNA data and called variants in 87 genes recurrently mutated in myeloid malignancies and/or MPN (supplemental Table 7). After applying RNA-specific filtering criteria (supplemental Methods: RNA-specific filter a-e), we identified 262 variants in 59 of 87 genes in 106 of 113 patients. Of the 262 variants, 221 were nonsynonymous SNVs, 13 were deletions, 18 were insertions, and 10 were stop-gain mutations (Figure 1C; supplemental Table 8).
To cross-validate variant calling based on RNA, 77 matching genomic DNA samples from the same patient were sequenced via the TruSight Myeloid Sequencing Panel (Illumina) (supplemental Table 9). We found an overall concordance of 82.2% for SNVs (10× coverage, variant allele frequency [VAF] > 0.1), which is consistent with published findings20,21 (supplemental Figure 6D; supplemental Table 10). To test whether VAFs vary significantly between RNA and DNA, we compared VAFs for JAK2-V617F assessed with allele-specific polymerase chain reaction on genomic DNA with VAFs obtained from RNA-seq. We observed a highly significant correlation between these 2 methods (R2 = 0.86, P = 2.2 × 10−16) (supplemental Figure 6C). In addition, most of the called RNA variants were called on clonal cell populations and were expressed in myeloid cells. More than half of the variants were annotated in the COSMIC database as “confirmed somatic mutation” and were frequently located in oncogenic hotspots (Figure 2; supplemental Figure 7; supplemental Tables 11 and 12).
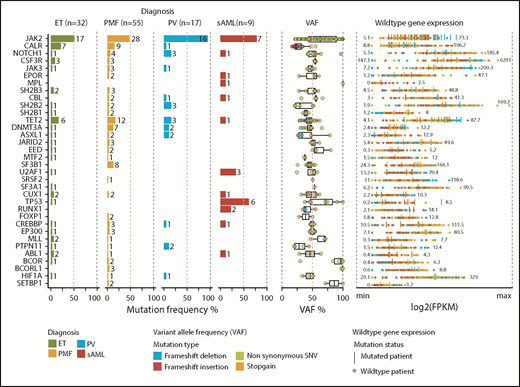
Recurrent gene mutations, VAF of mutations, and gene expression in MPN cohort. Thirty-five of 87 myeloid-related genes (Figure 1C; supplemental Table 7) with the highest mutation frequency within the cohort are depicted (left panel). Mutations are sorted as described in Figure 1C and colored by diagnosis. VAF for each mutation colored by mutation type (middle panel). Gene-expression values were reported for each patient as fragments per kilobase per million reads (FPKM) (supplemental Table 12) (right panel). Expression values are plotted as log2(FPKM). Patients with a mutation in the respective gene are shown as vertical bars. Numbers indicate the minimum and maximum expression values.
Recurrent gene mutations, VAF of mutations, and gene expression in MPN cohort. Thirty-five of 87 myeloid-related genes (Figure 1C; supplemental Table 7) with the highest mutation frequency within the cohort are depicted (left panel). Mutations are sorted as described in Figure 1C and colored by diagnosis. VAF for each mutation colored by mutation type (middle panel). Gene-expression values were reported for each patient as fragments per kilobase per million reads (FPKM) (supplemental Table 12) (right panel). Expression values are plotted as log2(FPKM). Patients with a mutation in the respective gene are shown as vertical bars. Numbers indicate the minimum and maximum expression values.
We identified a high frequency of mutations in epigenetic modifiers, such as TET2 (18.8% ET, 21.8% PMF, 17.7% PV) and DNMT3A (3.2% ET, 12.7% PMF, 11.8% PV), as well as an unexpectedly low frequency of ASXL1 mutations in PMF (3.13% ET, 3.64% PMF, 11.76% PV) compared with published data3,4 (Figure 2; supplemental Table 11). We also found unexpectedly high frequencies of mutations in NOTCH1 (3.1% ET, 7.3% PMF, 17.7% PV), SH2B2 (3.1% ET, 3.6% PMF, 17.7% PV), and SF3B1 (0% ET, 14.6% PMF, 0% PV).
SF3B1-mutated patients display a distinct pattern of 3′ splicing abnormalities
The high frequency of mutations in the SF3B1 gene in our RNA cohort was restricted to PMF patients (9/55; 16.3%). Furthermore, SF3B1 mutations were mutually exclusive with mutations in other genes of the splicing machinery (U2AF1, SRSF2, and SF3A1), and the patients were either JAK2-V617F positive or CALR-mutation positive (Figure 3A). The 9 PMF patients with SF3B1 mutations had nonsynonymous amino acid changes (K700E, K666N/R/T, and R594L) and, with the exception of R594L, all mutations coincided within mutation hotspot sites compared with mutation data extracted from the COSMIC database22 (Figure 3B). To evaluate whether these mutations introduce splicing abnormalities, we performed differential junction expression analysis comparing PMF patients carrying canonical SF3B1 mutations (K666/N/R/T and K700E) with the remaining 47 SF3B1 wild-type PMF patients and 15 healthy controls. Using DEXSeq, we identified 330 031 junctions, of which 850 differentially expressed junctions [adjusted P value < 0.1; log2(fold change) > 0] exhibited a novel 3′ss or 5′ss in the SF3B1-mutant patients (supplemental Table 13). Aberrant novel 3′ss were ∼4.9-fold more abundant than 5′ss (705 vs 145; Figure 3C). Focusing on aberrant 3′ss, we calculated the nucleotide distance for the aberrant 3′ss from the canonical splice site and observed a high enrichment between nucleotides 10 and 30 (Figure 3D). For 271 aberrant 3′ss within 10 to 30 nt from the canonical splice site, we could show distinct clustering of canonical (K700E, K666N/R/T) SF3B1 mutations and nonmutated or noncanonical SF3B1 (R594L) mutations (Figure 3E; supplemental Table 14). We observed shared and mutation-specific aberrant 3′ splicing patterns between K700E and K666N/R/T (Figure 3F). To examine whether other mutations in the splicing machinery (U2AF1, SRSF2, and SF3A1) have a similar impact on 3′ss, we included patients with mutations in these genes and performed hierarchical clustering. As previously reported for other cancer types,23 only K700E and K666N/R/T SF3B1 mutations showed a strong penetrance for the 3′ splicing abnormality phenotype (supplemental Figure 8). To examine whether the 3′ splicing abnormalities that we identified in PMF are shared with other malignancies, we compared differentially spliced junctions among 6 additional cancer types for which mutations in the splicing machinery have been described.24-27 This analysis revealed that the 3′ splicing abnormalities that we discovered in SF3B1-mutated PMF are disease specific. However, genes with the highest percentages of aberrant transcripts showed more frequent overlap among the analyzed cancer types (supplemental Figure 9). When we quantified the overlap across all cancer types, 2 genes with aberrant splicing (OXA1L and SLC3A2) were shared among all 7 analyzed cancer types (supplemental Figure 9C). In SF3B1-mutated PMF patients, we detected 141 unique splicing aberrations (supplemental Table 15).
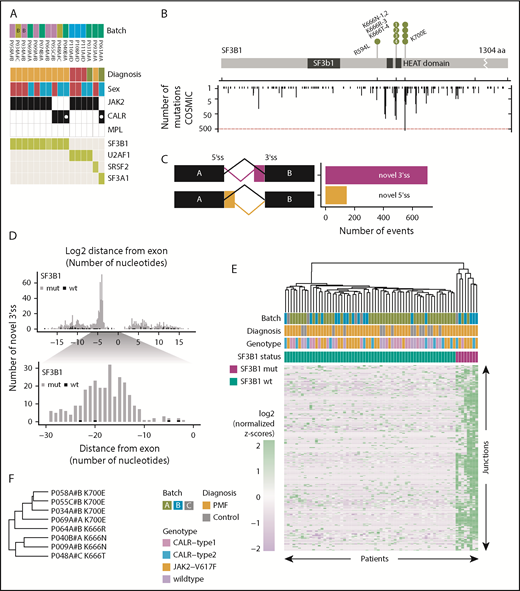
SF3B1-mutated PMF patients show distinct aberrant 3′ splicing. (A) SF3B1 mutations were present in 9 PMF patients, both JAK2- and CALR-positive patients. Other splicing factor mutations (U2AF1, SRSF2, and SF3A1) were mutually exclusive to SF3B1. (B) Gene mutation map of SF3B1. Nonsynonymous mutations in SF3B1 were located in oncogenic hotspot sites (K700E and K666N/R/T), with the exception of R594L. Mutations in SF3B1 were overlaid with mutation data from the COSMIC database22 (y-axis counts at the bottom). (C) Comparison of the frequencies of novel 3′ss and novel 5′ss. (D) Bar graphs showing the frequency of the log2 distance (in nucleotides) of the aberrant splice site from its canonical site. Zero indicates the start of the exon. (E) Hierarchical clustering of log2-normalized z-scores for 271 significantly upregulated 3′ss sites, located 10 to 30 bp away from the canonical splice site, in SF3B1+ patients (supplemental Table 14). (F) Mutation-specific splicing patterns were observed for amino acid changes K700 and K666.
SF3B1-mutated PMF patients show distinct aberrant 3′ splicing. (A) SF3B1 mutations were present in 9 PMF patients, both JAK2- and CALR-positive patients. Other splicing factor mutations (U2AF1, SRSF2, and SF3A1) were mutually exclusive to SF3B1. (B) Gene mutation map of SF3B1. Nonsynonymous mutations in SF3B1 were located in oncogenic hotspot sites (K700E and K666N/R/T), with the exception of R594L. Mutations in SF3B1 were overlaid with mutation data from the COSMIC database22 (y-axis counts at the bottom). (C) Comparison of the frequencies of novel 3′ss and novel 5′ss. (D) Bar graphs showing the frequency of the log2 distance (in nucleotides) of the aberrant splice site from its canonical site. Zero indicates the start of the exon. (E) Hierarchical clustering of log2-normalized z-scores for 271 significantly upregulated 3′ss sites, located 10 to 30 bp away from the canonical splice site, in SF3B1+ patients (supplemental Table 14). (F) Mutation-specific splicing patterns were observed for amino acid changes K700 and K666.
Identification of 3′ss with highest impact on predicted amino acid changes
Next, we aimed to identify those 3′ splice junction sites with the greatest impact on the protein sequence. We calculated the PSI value for each splice site and patient (Figure 4A), which represents the percentage of abnormal transcripts in the total number of transcripts. A total of 43 novel 3′ splice junctions showed a median PSI 20% for SF3B1-mutated patients (supplemental Table 16). After predicting the impact of each splicing abnormality on the amino acid sequence, we selected 20 splicing abnormalities that resulted in a frameshift mutation and truncation of the protein or introduced a stretch of novel amino acids in frame to the native protein sequence. Hierarchical clustering of PSI values for aberrant 3′ss in relation to canonical splice sites showed preferential usage of aberrant 3′ss by SF3B1-mutated patients compared with SF3B1 wild-type patients (Figure 4B). Aberrant 3′ss usage was also dependent on specific SF3B1 mutants (K700 and K666) (Figure 4C). We confirmed the presence of 19 of 20 abnormal splice events detected by RNA-seq using an alternative methodology (fragment length analysis of reverse-transcription polymerase chain reaction products) in 4 patients (P009A, P040B, P048A, P069A) (Figure 4D-E). Several attributes for the selected 20 genes are summarized in Figure 4F. Interestingly, of the 16 out-of-frame truncations, only the TTI1 gene showed a significant [log2(fold change) = −0.38; adjusted P value = .034] decrease in the messenger RNA (mRNA level), suggesting that non-sense–mediated decay does not influence the transcript abundance in most of the selected genes. Moreover, OXA1L even showed a significant elevation [log2(fold change) = 0.86; adjusted P value = 2.02 × 10−7] in the mRNA level, despite the presence of a truncation (supplemental Table 17).
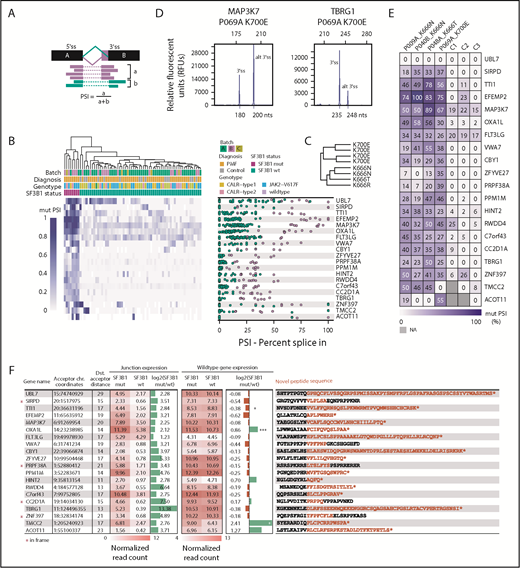
Top 20 genes, ranked by highest PSI value, have putative alterations on the protein level. (A) PSI values for alternative 3′ss were calculated by dividing the number of reads spanning the alternative 3′ss by the number of reads spanning the alternative 3′ss + the number of reads spanning the canonical splice site. (B) Hierarchical clustering of PSI values of novel 3′ss vs canonical 3′ss of the top 20 genes ranked by ascending PSI values. Only junction sites introducing novel peptide sequences were considered. (C) PSI-specific splicing patterns were observed for amino acid changes K700 and K666. (D) Fragment length analysis was used to validate RNA-based PSI measurements. (E) Validation of PSI values for 4 patients and 3 healthy controls with fragment length analysis. Experimental PSI values were calculated using relative peak heights of canonical and alternate 3′ss. No alternative 3′ splicing for UBL7 was present in patients or healthy controls. (F) Summary of differential splice junction (junction expression, columns 4 and 5) and gene expression analysis (wild-type gene expression, columns 7 and 8) of SF3B1 mutated versus wild-type patients. Differential splice junction and gene expression identified 850 and 828 significantly regulated junctions and genes, respectively (supplemental Tables 13 and 17). Normalized junction and gene read counts were extracted for the previously identified top 20 genes with highest PSI. Columns 6 and 9: log2(fold change) mutant vs wild-type for junction and gene expression. Column 10: predicted amino acid sequences introduced by the splicing defects. *P < .05, ***P < .0005 (adjusted P values). chr., chromosome; Dst., distal (distal acceptor distance − number or nucleotides between aberrant and canonical splice site); mut, mutant; wt, wild-type.
Top 20 genes, ranked by highest PSI value, have putative alterations on the protein level. (A) PSI values for alternative 3′ss were calculated by dividing the number of reads spanning the alternative 3′ss by the number of reads spanning the alternative 3′ss + the number of reads spanning the canonical splice site. (B) Hierarchical clustering of PSI values of novel 3′ss vs canonical 3′ss of the top 20 genes ranked by ascending PSI values. Only junction sites introducing novel peptide sequences were considered. (C) PSI-specific splicing patterns were observed for amino acid changes K700 and K666. (D) Fragment length analysis was used to validate RNA-based PSI measurements. (E) Validation of PSI values for 4 patients and 3 healthy controls with fragment length analysis. Experimental PSI values were calculated using relative peak heights of canonical and alternate 3′ss. No alternative 3′ splicing for UBL7 was present in patients or healthy controls. (F) Summary of differential splice junction (junction expression, columns 4 and 5) and gene expression analysis (wild-type gene expression, columns 7 and 8) of SF3B1 mutated versus wild-type patients. Differential splice junction and gene expression identified 850 and 828 significantly regulated junctions and genes, respectively (supplemental Tables 13 and 17). Normalized junction and gene read counts were extracted for the previously identified top 20 genes with highest PSI. Columns 6 and 9: log2(fold change) mutant vs wild-type for junction and gene expression. Column 10: predicted amino acid sequences introduced by the splicing defects. *P < .05, ***P < .0005 (adjusted P values). chr., chromosome; Dst., distal (distal acceptor distance − number or nucleotides between aberrant and canonical splice site); mut, mutant; wt, wild-type.
RNA-based workflow for systematic identification of neoantigens
Published workflows for neoantigen discovery rely on exome sequencing combined with transcriptome sequencing to focus only on expressed variants.28-30 In our approach, we bypassed the exome sequencing step by calling the expressed mutation classes directly from transcriptome data. In addition, we called MHCI haplotypes directly from transcriptome data for 113 MPN patients and controls (supplemental Table 18). For each of the HLA-A, HLA-B, and HLA-C genes, we identified the 4 most frequent alleles in our cohort (supplemental Table 19). Next, we used the peptide-to-MHCI (peptide:MHCI complex) binding prediction algorithm NetMHCpan19 to predict tumor-specific neoantigens for MHCI presentation.
To examine the number of predicted neoantigens for the 12 most common HLA alleles in our patient cohort, we generated a virtual peptide library based on the mutations detected at the RNA level. This peptide library consisted of 541 patient-specific (or 149 unique) peptides predicted to have a binding affinity for 1 of the 12 common MHCI proteins. At the cohort level, 102 were predicted to have a strong binding (%Rank < 0.5), whereas 439 were weak binders (%Rank < 2) (Figure 5A; supplemental Table 20). In 86% of peptide:MHCI pairs, the peptide was derived from frameshift-causing mutations, emphasizing their potential role as a source of neoantigens (supplemental Figure 10). Although CALR occupied the majority (n = 403; 86.9%) of peptide:MHCI pairs, neoantigens derived from frameshift mutations in TET2 (n = 16; 3.5%) were also present, but none of these were shared within the MPN cohort.
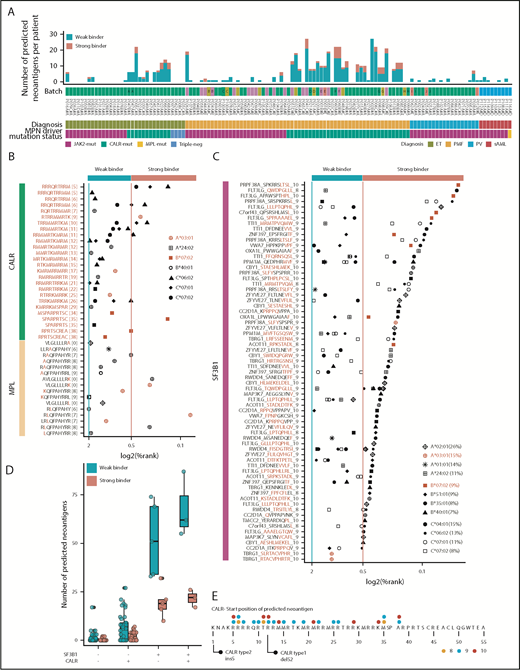
MPN driver mutations (CALR and MPL) and SF3B1-mediated splicing aberrations are a rich source of predicted neoantigens. (A) Number of predicted neoantigens (nonsynonymous SNVs, indels, and fusions) for each patient, based on individual HLA haplotypes. For prediction, the 4 most common alleles for HLA-A, HLA-B, and HLA-C within the MPN cohort were considered (supplemental Table 19). Thresholds for weak binders (%Rank < 2 and >0.5) and strong binders (%Rank < 0.5) were selected based on the NetMHCpan authors’ recommendation. Percentile rank score (%Rank) values for CALR and MPL (B) and SF3B1 peptides predicted with NetMHCpan (C) are plotted. HLA alleles, marked in red, were used for validation (Figure 6). HLA allele frequencies for the MPN cohort are indicated in panel C. (D) Number of predicted neoantigens (all mutation classes combined) for each patient with no CALR or SF3B1 mutation, 1 of the 2 mutations, or both mutations, separated into weak and strong binders. CALR-mutated patients had a mean of 8.0 weak binders and 2.3 strong binders, whereas SF3B1-mutated patients, without a cooccurring CALR mutation, had an average of 38.2 weak binders and 16.2 strong binders. For JAK2-V617F–mutated patients, we found an average of 0.9 and 0.1 predicted neoantigens for weak and strong binders, respectively (supplemental Table 23). (E) CALR mutant tail “consensus sequence” as reported by Klampfl et al.1 The dots indicate the starting positions of peptides colored by peptide length.
MPN driver mutations (CALR and MPL) and SF3B1-mediated splicing aberrations are a rich source of predicted neoantigens. (A) Number of predicted neoantigens (nonsynonymous SNVs, indels, and fusions) for each patient, based on individual HLA haplotypes. For prediction, the 4 most common alleles for HLA-A, HLA-B, and HLA-C within the MPN cohort were considered (supplemental Table 19). Thresholds for weak binders (%Rank < 2 and >0.5) and strong binders (%Rank < 0.5) were selected based on the NetMHCpan authors’ recommendation. Percentile rank score (%Rank) values for CALR and MPL (B) and SF3B1 peptides predicted with NetMHCpan (C) are plotted. HLA alleles, marked in red, were used for validation (Figure 6). HLA allele frequencies for the MPN cohort are indicated in panel C. (D) Number of predicted neoantigens (all mutation classes combined) for each patient with no CALR or SF3B1 mutation, 1 of the 2 mutations, or both mutations, separated into weak and strong binders. CALR-mutated patients had a mean of 8.0 weak binders and 2.3 strong binders, whereas SF3B1-mutated patients, without a cooccurring CALR mutation, had an average of 38.2 weak binders and 16.2 strong binders. For JAK2-V617F–mutated patients, we found an average of 0.9 and 0.1 predicted neoantigens for weak and strong binders, respectively (supplemental Table 23). (E) CALR mutant tail “consensus sequence” as reported by Klampfl et al.1 The dots indicate the starting positions of peptides colored by peptide length.
MPN driver mutations CALR and MPL are a rich source of neoantigens
Driver mutations in MPN are well characterized and are represented by JAK2-V617F, MPL-W515K/L/A, and a frameshift mutation in CALR. Because the more frequent MPL-W515L and MPL-W515K point mutations were not present in our cohort, we tested these separately for peptide:MHCI interactions. Predictions of 4 of the most common HLA-A, HLA-B, and HLA-C alleles resulted in 42 peptide:MHCI pairs for CALR and 17 peptide:MHCI pairs for MPL-W515K/L/A (Figure 5B; supplemental Tables 20 and 21). The CALR ins5 mutation forms more unique predicted neoantigens than the CALR del52 mutation toward the N terminus of the CALR protein (Figure 5E). We did not identify any binding peptides derived from the JAK2-V617F driver mutation for any of the HLA genotypes tested.
Identification of neoepitopes in aberrantly spliced genes
Next, we wanted to examine whether the peptide sequences derived from abnormally spliced genes in SF3B1-mutated patients may serve as putative neoantigens. Of the 850 differentially expressed junctions, 21 aberrant splice sites (supplemental Table 13) were not expressed in SF3B1 wild-type patients. Although these 21 splice sites may, from a tumor-specificity point of view, present suitable candidates for neoantigens derived from aberrant splice sites, none of them met the other equally important criteria defined in supplemental Methods. Therefore, we focused our analysis on our previously defined set of 19 genes affected by aberrant splicing listed in Figure 4F, leaving the question of true tumor specificity open. We used NetMHCpan to predict peptide:MHCI binding, applying the same criteria for weak and strong binders as previously described. A total of 169 peptides had ≥1 MHCI protein with a predicted binding %Rank < 2 (Figure 5C; supplemental Table 22). We counted the number of predicted neoantigens considering each patient’s personal MHCI haplotype. Patients with SF3B1 and CALR co-occurring mutations had the highest number of neoantigens presented by the patient’s own MHCI variants (a mean of 68.0 predicted weak binders and a mean of 21.6 predicted strong binders) (Figure 5D; supplemental Table 23).
In vitro validation of predicted peptides to MHCI protein
An ELISA-based peptide-exchange assay was used to validate the predicted peptide:MHCI binding.31 We tested 35 peptides predicted to bind weakly (n = 15) or strongly (n = 20) to 4 commercially available HLA monomers (A*03:01, A*11:01, B*07:02, and B*08:01). Control peptides of known strong or weak binding affinity were available for A*03:01, A*11:01, and B*07:02 (Figure 6; supplemental Table 24).We defined a threshold for binders vs nonbinders as 2 standard deviations from the mean of the absorbance values of 3 positive-control peptides in the ELISA assay. Using an in vitro binding assay, we could validate 23 of the 35 (65.7%) predicted peptide:MHCI interactions.
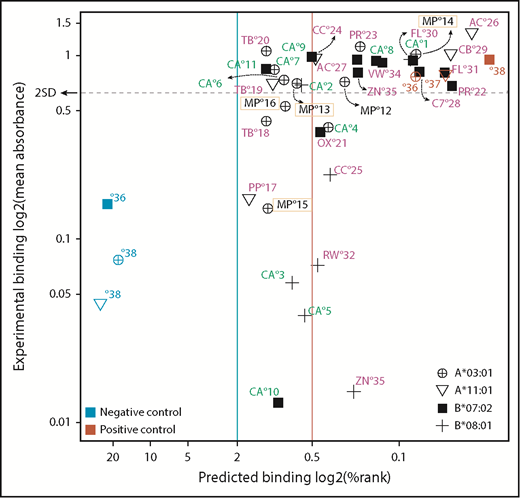
In vitro validation of NetMHCpan predictions. Selected peptides (supplemental Table 24) were synthesized and tested for binding to 4 MHCI molecules using an ELISA-based peptide-exchange assay. The threshold for predicted binding was set as described for Figure 5A. For experimental binding validation, the threshold was set at 2 standard deviations (2SD) from the mean absorbance value for 3 positive-control peptides (colored in red). Peptides above this threshold were considered strong binders. Of the 20 predicted strong binders, 15 exhibited binding values above the threshold (75.0%). Of the 15 predicted weak binders, 8 were above the threshold (53.3%). Color coding: green, CALR-derived peptides; yellow rectangle, MPL-derived peptides; magenta, SF3B1-derived peptides. Abbreviations are listed in supplemental Table 24.
In vitro validation of NetMHCpan predictions. Selected peptides (supplemental Table 24) were synthesized and tested for binding to 4 MHCI molecules using an ELISA-based peptide-exchange assay. The threshold for predicted binding was set as described for Figure 5A. For experimental binding validation, the threshold was set at 2 standard deviations (2SD) from the mean absorbance value for 3 positive-control peptides (colored in red). Peptides above this threshold were considered strong binders. Of the 20 predicted strong binders, 15 exhibited binding values above the threshold (75.0%). Of the 15 predicted weak binders, 8 were above the threshold (53.3%). Color coding: green, CALR-derived peptides; yellow rectangle, MPL-derived peptides; magenta, SF3B1-derived peptides. Abbreviations are listed in supplemental Table 24.
Discussion
We studied the global transcriptome of 113 MPN patients using RNA-seq technology and described their mutational landscapes in detail. We established an RNA-seq–based pipeline utilizing high-quality transcriptome data as an alternative method for neoantigen discovery. This is the first study to globally annotate the MPN transcriptome with the aim to identify and systematically mine MPN-relevant putative neoantigens for targeted immunotherapy of MPN.
Another novel aspect of this study is the global analysis of gene fusions in MPN that are missed by targeted or exome sequencing. We detected and annotated gene fusions in a systematic and unbiased high-throughput transcriptome-wide approach. Fusion genes found in chronic MPN (JAK2 or CALR positive) were scarce. With the exception of known and in-frame BCR-ABL1 oncogenic fusion found in 1 PMF patient, 10 fusions detected in chronic MPN were novel and out of frame. For some fusions, we observed high expression of the wild-type gene and low expression of the fusion transcript, which are suggestive of fusion formation through trans-splicing events, as suggested by previous studies.32,33 Lack of recurrence for any of the identified fusions prohibited any mechanistic studies. Two of 9 post-MPN sAML patients had fusion genes (INO80D-GPR1 and XPO5-RUNX2) that have not been previously described. Surprisingly, no typical de novo acute myeloid leukemia fusion oncogenes were detected, emphasizing the distinct nature of disease progression in post-MPN sAML compared with de novo acute myeloid leukemia.34 Interestingly, predicted protein products of fusion genes, in particular frameshift mutations or their junction regions, were not a significant source of neoantigens.
We focused our analysis on 86 genes frequently mutated in clonal myeloid diseases. In addition, we reported HLA genotypes extracted from RNA-seq data in a cohort of 113 MPN patients. This enabled us to make a truly personalized prediction of neoantigen occurrence for each MPN patient. Although the numbers of indels and SNVs identified per patient were low, as expected for MPN,4 we could still identify 149 unique neoantigens in 62% of MPN patients that might have potential use for personalized cancer immunotherapy approaches.
Having defined a gene set with the highest impact on protein sequence, we hypothesized that splicing abnormalities induced by canonical SF3B1 mutations may serve as potential neoantigens for MPN. Several findings strengthened our assumptions that frameshift mutations caused by aberrant splicing might indeed serve as potent neoantigens. We could show high expression and high VAF of the mutant copy of SF3B1. The aberrantly spliced genes that we described did not show frequent downregulation at mRNA levels (with exception of TTI1), arguing against the involvement of non-sense–mediated decay of aberrant transcripts, as previously suggested by other studies.26,35 Furthermore, for 15 peptides derived from abnormally spliced genes, we showed a strong affinity to common MHCI molecules using an in vitro binding assay. Although additional studies are needed to demonstrate their immunogenicity, peptides derived from splicing defects may represent recurrent immunotherapy targets, perhaps as valuable as peptides derived from the frameshifted CALR oncogenic MPN driver. Finally, although our research did not provide evidence of these aberrant transcripts at the protein level, a recent study identified so-called “neojunctions” in SF3B1-mutated cancers and confirmed the presence of these neojunctions at the protein level using mass spectrometry approaches.39 We believe that it would be of great interest for future research to formally show that our candidate splicing-derived putative neoantigens are indeed translated to proteins and, therefore, are potentially capable of being presented in MHCI complexes on the cell surface.
Another important question is the tumor specificity of the splicing defects. Aberrant splicing at a specific junction may also occur at a lower frequency in healthy cells so that targeting the resulting protein sequence may cause autoimmunity and toxicity.37,38 For several candidate genes, our results show a weak presence of the aberrant transcripts in SF3B1-nonmutated patients. Nevertheless, a search through the human proteome database with the translated novel protein sequences did not yield any significant matches, excluding the possibility that any of these aberrant transcripts are merely alternative isoforms. Thus, further experimental evidence to show tumor specificity at the protein level is required to classify whether aberrant 3′ss are true neoantigens or tumor-associated antigens overexpressed in tumor cells.
Our future studies will be aimed at annotating the candidate neoantigenic peptides identified in this study for the ability to induce T-cell activation in healthy controls and in SF3B1-mutated MPN patients to prove that these are truly immunogenic antigens. In summary, our study may serve as a foundation and resource for the development of individualized vaccines or adoptive cell-based therapies for MPN, in particular for PMF patients, in whom the unmet medical need is greatest.
Whole-transcriptome sequence data have been deposited in the European Genome-phenome Archive under accession number EGAS00001003486.
The source code for the implementation of the workflows described in this study is available for download and use on GitHub (http://github.com/sp00nman/rnaseqlib).
The online version of this article contains a data supplement.
The publication costs of this article were defrayed in part by page charge payment. Therefore, and solely to indicate this fact, this article is hereby marked “advertisement” in accordance with 18 USC section 1734.
Acknowledgments
The authors thank all the members of the Kralovics Laboratory, Johanna Klughammer, and Ilaria Casetti for discussion and feedback. The authors also thank the Biomedical Sequencing Facility at CeMM for RNA sequencing.
This work was supported by Austrian Science Fund Projects F4702-B20 and P29018-B30 (studies performed in Vienna). Studies conducted at the University of Pavia were supported by Associazione Italiana per la Ricerca sul Cancro, Milan, Italy (AIRC 5x1000 call “Metastatic disease: the key unmet need in oncology,” project 21267; a detailed description of the MYNERVA project is available at http://www.progettoagimm.it).
Authorship
Contribution: F.S. and R.K. conceived and designed the experiments and wrote the manuscript; F.S., R.K., R.J., F.R., E.H., R.H., E.F., J.D.M.F., and E.B. performed the research and experiments; F.S., M. Schuster, J.D.M.F., C.B., and R.K. analyzed and interpreted the data and performed statistical analysis; J.D.M.F., B.G., M. Schalling, E.R., D.P., G.F., I.F., L.V., J.M., T.H., M.M., A.S., M.C., and H.G. contributed materials and analysis tools. All authors contributed to the final version of the manuscript.
Conflict-of-interest disclosures: R.K. has received honoraria and/or research funding from PharmaEssentia, AOP Orphan Pharmaceuticals, and Novartis; has acted as a consultant for PharmaEssentia, AOP Orphan Pharmaceuticals, and QIAGEN; and owns stock in MyeloPro Diagnostics and Research. The remaining authors declare no competing financial interests.
Correspondence: Robert Kralovics, CeMM Research Center for Molecular Medicine of the Austrian Academy of Sciences, Lazarettgasse 14, AKH BT25.3, 1090 Vienna, Austria; e-mail: rkralovics@cemm.oeaw.ac.at.

This feature is available to Subscribers Only
Sign In or Create an Account Close Modal