

In this issue of Blood, identify aberrant activation of inflammatory signaling pathways as a result of the Hippo kinase, MST1, loss in myeloid malignancies with deletion of chromosome 20q.1
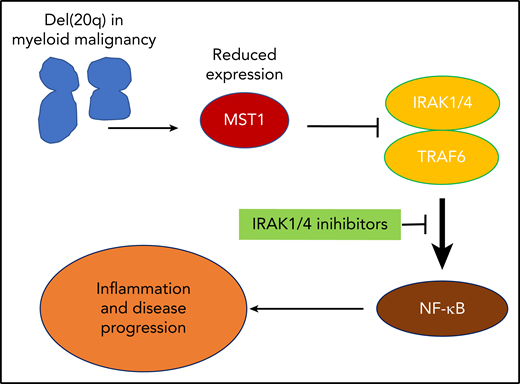
Reduced expression of MST1 in del(20q) leads to pathological activation of NF-κB signaling and downstream inflammatory pathways. This identifies a novel treatment pathway through the inhibition of upstream mediators IRAK1/4.
Reduced expression of MST1 in del(20q) leads to pathological activation of NF-κB signaling and downstream inflammatory pathways. This identifies a novel treatment pathway through the inhibition of upstream mediators IRAK1/4.
Chromosomal aberrations are a common feature of many cancer subtypes. Recurring cytogenetic abnormalities that are found specifically in particular cancers may provide important information about disease pathogenesis and identify a path toward treatment susceptibility.2 Deletions in the long arm of chromosome 20, or del(20q), is a common cytogenetic finding in patients with blood cancers, particularly myelodysplasia, myeloproliferative neoplasm (MPN), and acute leukemia. Interestingly, when found in patients with myelodysplasia (MDS), it is associated with a relatively favorable prognosis3 and contrasts with the adverse prognostic effect of other cytogenetic abnormalities such as monosomy of chromosome 7 or chromosome 3p abnormalities. In MPN, cytogenetic abnormalities are associated with more advanced disease, and del(20q) is the most common chromosomal aberration in primary and postpolycythemic myelofibrosis.4 Mapping of the commonly deleted region of chromosome 20q has identified a number of candidate genes that may contribute to myeloproliferative neoplasm with haploinsufficient expression.5
In the accompanying manuscript, Stoner et al examined gene expression changes from patients with del(20q) MDS and found that many of the downregulated genes were located on del(20q). In particular, STK4, encoding the Hippo kinase MST1, was downregulated in MDS with del(20q) and myelofibrosis. Functionally, deletion of STK3 and STK4 Hippo kinases in normal hematopoietic cells induced an MDS-like state characterized by thrombocytopenia, enlarged platelets, and myeloid skewing. These effects were also seen at the level of hematopoietic stem cells, as evidenced by loss of hematopoietic stem cell fitness and engraftment. This was dependent on gene dosage, with the most pronounced findings in homozygous deletion. Furthermore, retroviral expression of human JAK2V617F cooperated with STK3/4 haploinsufficiency to increase the rate of myelofibrosis and disease progression. Again, these findings reflect the association of del(20q) and other chromosomal changes with progression of MPN from early stage disease, such as polycythemia vera, to advanced myelofibrosis.
Mechanistically, the progression to myelofibrosis was associated with inflammatory cytokines, in particular increased expression of interleukin 6 (IL-6), IL-1β, IL-15, and MMP-3. Using pathway analysis of gene expression studies, they were able to show that this mutant JAK2V617F and STK3/4 haploinsufficient context had activated regulators of innate immune signaling and inflammatory regulators associated with activation of the NF-κB pathway. To understand how MST1 loss might lead to NF-κB activation, the authors concentrated on the described interaction between MST1 and IRAK1 and confirmed this finding, while also showing that MST1 expression was able to suppress IRAK-TRAF6 mediated activation of NF-κB signaling and that this suppression was dependent on the kinase activity of MST1.
Activation of the NF-κB pathway has been found in other models of secondary myelofibrosis,6 and targeting this pathway has some efficacy in preventing the progression of disease.7 Therefore, to translate these findings, the authors used an IRAK1/4 inhibitor in the JAK2V617F, Stk3/4 haploinsufficient mouse model of myelofibrosis. This proinflammatory signaling that characterized the myelofibrotic phenotype was inhibited by the IRAK1/4 inhibitor in vivo, providing a logical path to clinical translation that may benefit patients with advanced MPN and increased inflammatory signaling (see figure).
Altogether, Stoner et al provide a compelling argument for a functional role of MST1 loss in the pathogenesis of myeloid malignancies, leading to dysregulated inflammatory signaling and progression to advanced disease. A number of questions still remain, such as identifying the best way of dampening down inflammatory signaling in these diseases and why del(20q) leads to a relatively favorable prognosis compared with other cytogenetic abnormalities. This work builds on a recurrent theme that identifies pathological activation of inflammation and immune pathways in myeloid blood cancers and provides hope that these findings may be leveraged to design new treatments for our patients with these diseases.
Conflict-of-interest disclosure: The author declares no competing financial interests.

This feature is available to Subscribers Only
Sign In or Create an Account Close Modal