

Key Points
IRF4 deletion in Tcl-1 tg mice and IRF4low CLL patients enhances disease progression due to increased tumor immune evasion.
This is caused by a downregulation of the antigen processing and presentation machinery and reduced T-cell costimulation.

Abstract
Chronic lymphocytic leukemia (CLL) is a heterogenous disease that is highly dependent on a cross talk of CLL cells with the microenvironment, in particular with T cells. T cells derived from CLL patients or murine CLL models are skewed to an antigen-experienced T-cell subset, indicating a certain degree of antitumor recognition, but they are also exhausted, preventing an effective antitumor immune response. Here we describe a novel mechanism of CLL tumor immune evasion that is independent of T-cell exhaustion, using B-cell–specific deletion of the transcription factor IRF4 (interferon regulatory factor 4) in Tcl-1 transgenic mice developing a murine CLL highly similar to the human disease. We show enhanced CLL disease progression in IRF4-deficient Tcl-1 tg mice, associated with a severe downregulation of genes involved in T-cell activation, including genes involved in antigen processing/presentation and T-cell costimulation, which massively reduced T-cell subset skewing and exhaustion. We found a strong analogy in the human disease, with inferior prognosis of CLL patients with low IRF4 expression in independent CLL patient cohorts, failed T-cell skewing to antigen-experienced subsets, decreased costimulation capacity, and downregulation of genes involved in T-cell activation. These results have therapeutic relevance because our findings on molecular mechanisms of immune privilege may be responsible for the failure of immune-therapeutic strategies in CLL and may lead to improved targeting in the future.
Introduction
Chronic lymphocytic leukemia (CLL) accounts to 25% to 30% of all leukemias in Western countries, with incidence rates ranging from 3.65 to 6.75 cases per 100 000 population per year.1,2 CLL is characterized by an outgrowth of malignant CD19/CD5 double positive B cells, mainly residing in the peripheral blood, bone marrow, and the lymphoid organs, and by a high biologic heterogeneity reflected in clinically different outcomes including disease progression, therapy response, and relapse.3,4 Microenvironmental signals contribute to this heterogeneity and are derived from either the stromal cell compartment or components of the immune system that include (auto)antigens, B-cell receptor signaling, monocytes, macrophages, and T cells.5-9 T cells from CLL patients are skewed from the naïve to the memory T-cell compartment and thus represent an activated and potentially antigen and/or tumor experienced T-cell subset.10,11 The functionality of these T cells, however, is impaired by the elevated expression of exhaustion markers and by defects in the formation of immunological synapses.12-14 Analogous defects in T cell–mediated antitumor immunity were also observed in Tcl-1 tg mice,12,14-17 which develop a murine CLL with late onset and high penetrance.18 Using this model, we and others established that the CLL typical T-cell skewing was directly induced by CLL tumor cells,14,15 supporting the hypothesis of a tumor-specific transcriptional program that is active in CLL cells that favors CLL tumor immune evasion by manipulating the CLL cell cross talk with other components of the immune system. The mechanisms that establish and retain immune evasion and alter gene transcription in CLL tumor cells are, however, still poorly understood.
One potential candidate transcription factor is interferon regulatory factor 4 (IRF4), which controls the differentiation of B, T, dendritic, and myeloid cells in a context-dependent manner and regulates various aspects relevant for a functional immune response.19 In T cells, IRF4 is crucial for T-cell differentiation and expansion,20-24 in dendritic cells IRF4 contributes to the regulation of antigen presentation,25,26 promotes macrophage differentiation, and blocks the generation of myeloid-derived suppressor cells.27-29 In B cells, IRF4 regulates B-cell receptor signaling30 ; contributes in class switch recombination, somatic hypermutation, and germinal center response; and is essential for plasma cell development.31-33 IRF4 is also involved in cell proliferation and survival and described as an oncogene in multiple myeloma and some subtypes of DLBCL.34,35 By contrast, tumor-suppressive functions were observed in pre-B-cell leukemias and in c-Myc–induced malignancies.36-38 In CLL single nucleotide polymorphisms in the IRF4 gene, were related to CLL susceptibility and in next-generation sequencing, mutations in the IRF4 gene were detected in patients with overt disease.39-42 Germline deficiency of IRF4 in the mouse model was associated with severe defects in T-cell cytotoxicity and antitumor response.33 Therefore, we generated Tcl-1 tg mice with B cell–specific IRF4 deletion and are the first to describe enhanced CLL disease progression by tumor immune evasion in this mouse model and validated these data also in the human disease.
Materials and methods
Mouse strains and CLL patient material
Murine CLL samples are derived from Eµ-Tcl1 transgenic (tg) mice18 and crossed to CD19Cre IRF4flox/flox mice.32 For adoptive transfer 10 × 106 splenocytes were injected intraperitoneal into C57/BL6 or NOD-SCID recipients.43 Human CLL blood samples were collected at the IIIrd Medical Department (University Hospital Salzburg, Austria) upon written informed consent in accordance with the Declaration of Helsinki. Patient details are summarized in supplemental Table 1, available on the Blood Web site. All studies in mice were approved by the Austrian Federal Ministry of Education, Science and Research. All studies on patient-derived material were approved by the Salzburg ethics committee.
Immune phenotyping, single-cell mass cytometry, and cell preparation
Immune phenotyping was performed on a Gallios device (Beckman Coulter) and single-cell mass cytometry on a Helios device (CyTOF, Fluidigm). Antibodies used for flow cytometry and single-cell mass cytometry are summarized in supplemental Table 2. CLL cell purification, RNA isolation, B-cell receptor clonality analysis, and in vitro culture assays were performed as described.43-45
RNA-Seq and Affymetrix GeneChip analysis
Supplementary methods
Further details on sample preparation, experimental conditions, and bioinformatic analysis workflows are provided in the supplemental Methods.
Results
B cell–specific IRF4 deletion in Tcl-1 tg mice accelerates CLL disease development
To analyze a potential role of B cell–specific IRF4 expression in CLL pathogenesis, we crossed IRF4 floxed,32 CD19 Cre, and Tcl-1 tg mice18 to generate Tcl-1 tg mice with homozygous IRF4 deletion specifically in the B-cell lineage (Tcl-1 tg IRF4 ΔB/ΔB) in comparison with Tcl-1 tg mice with wild-type IRF4 expression (Tcl-1 tg IRF4+/+). As a first approach, we studied the development of CLL over time (Figure 1A). As shown in Figure 1B, IRF4 ΔB/ΔB accumulated CD19+ CD5+ CLL cells earlier and disease progression was faster. This translated to shorter overall survival times of Tcl-1 tg IRF4 ΔB/ΔB mice (7.5 months in median) as compared with Tcl-1 tg IRF4+/+ littermates (11.9 months in median) (Figure 1C). At time of death, splenomegaly was observed in both genotypes (Figure 1D, green bars). The spleen weight was not different between the genotypes, whereas the spleen size was slightly increased in Tcl-1 tg IRF4+/+ mice (Figure 1D). Thus, we next analyzed the percentage of infiltrating CLL cells in all lymphoid organs at time of death. Organ infiltration of CLL cells was similar in Tcl-1 tg IRF4+/+ and Tcl-1 tg IRF4 ΔB/ΔB littermates (Figure 1E). Because IRF4 plays important roles in B-cell differentiation, we verified the phenotype of CLL or B cells and observed no differences in surface marker expression (supplemental Figure 1A-D). The lack of IRF4 per se was not a tumor driver because 24/26 IRF4 ΔB/ΔB littermates without Tcl-1 tg remained healthy (supplemental Figure 1E), did not accumulate CLL cells in the peripheral blood (supplemental Figure 1F) or other organs (supplemental Figure 1G), and spleen enlargement was not observed (Figure 1D). Because Tcl-1 tg mice develop B-cell receptor (BCR) rearranged clones with varying degrees of oligoclonality,43 we investigated the clonal structures by genotype, but observed no differences (Figure 1F), indicating similar clonal selection pressures, irrespective of the IRF4 status.
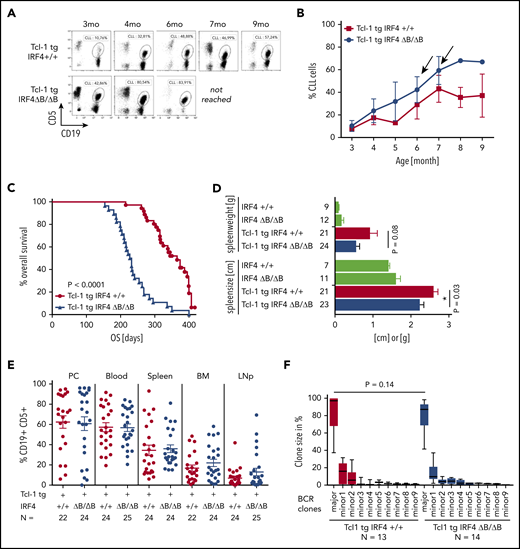
CLL development in Tcl-1 tg mice with and without IRF4 expression. (A) CD19+ CD5+ CLL cells were measured in the peripheral blood using flow cytometry and (B) analyzed over time. N = 6 (Tcl-1 tg IRF4+/+, red), N = 8 (Tcl-1 tg IRF4 ΔB/ΔB, blue). Arrows indicate mice euthanized because of overt CLL development. (C) Kaplan-Maier analysis showing overall survival of Tcl-1 tg IRF4+/+ mice (N = 36) and Tcl-1 tg IRF4 ΔB/ΔB mice (N = 28). The study end point of this analysis was 400 ± 20 days. (D) Spleen size and weight were analyzed in healthy age-matched littermates of the indicated genotypes (green) and leukemic Tcl-1 tg mice with WT IRF4 expression (red) or without IRF4 expression (blue). Analyzed mouse numbers are depicted on the y-axis. (E) CD19+ CD5+ CLL cells were measured in lymphoid organs derived from leukemic Tcl-1 tg mice with WT IRF4 expression (red) or without IRF4 expression (blue). (F) CLL clone sizes as measured by BCR sequencing. LNp, peripheral lymph nodes; major, major clone in % of total CLL cells; minor, minor clones in % of CLL cells; PC, peritoneal cavity; WT, wild-type.
CLL development in Tcl-1 tg mice with and without IRF4 expression. (A) CD19+ CD5+ CLL cells were measured in the peripheral blood using flow cytometry and (B) analyzed over time. N = 6 (Tcl-1 tg IRF4+/+, red), N = 8 (Tcl-1 tg IRF4 ΔB/ΔB, blue). Arrows indicate mice euthanized because of overt CLL development. (C) Kaplan-Maier analysis showing overall survival of Tcl-1 tg IRF4+/+ mice (N = 36) and Tcl-1 tg IRF4 ΔB/ΔB mice (N = 28). The study end point of this analysis was 400 ± 20 days. (D) Spleen size and weight were analyzed in healthy age-matched littermates of the indicated genotypes (green) and leukemic Tcl-1 tg mice with WT IRF4 expression (red) or without IRF4 expression (blue). Analyzed mouse numbers are depicted on the y-axis. (E) CD19+ CD5+ CLL cells were measured in lymphoid organs derived from leukemic Tcl-1 tg mice with WT IRF4 expression (red) or without IRF4 expression (blue). (F) CLL clone sizes as measured by BCR sequencing. LNp, peripheral lymph nodes; major, major clone in % of total CLL cells; minor, minor clones in % of CLL cells; PC, peritoneal cavity; WT, wild-type.
IRF4 deleted Tcl-1 tg tumors show enhanced tumor immune evasion
Given the function of IRF4 as transcription factor, we next performed RNA-Seq in purified CLL cells derived from the spleen of 4 Tcl-1 tg IRF4+/+ and 3 Tcl-1 tg IRF4 ΔB/ΔB tumors. By differential gene expression analysis, we found 1530 deregulated genes with a false discovery rate–corrected P value <.05 and a log2 fold-change of at least ± 1.5 (supplemental Table 3). A gene ontology (GO) enrichment analysis was applied to identify functionally related gene clusters, and we found a significant enrichment of 19 biological process GO terms (supplemental Table 4). Genes upregulated in IRF4-deficient mice were mainly correlated to GO terms associated with cell-cycle processes (Figure 2A, red bars). By contrast, downregulated genes could be directly or indirectly linked to immune response processes (Figure 2A, green bars; supplemental Figure 2). Despite the restriction of this analysis to sorted B cells, the highest statistical significance in the entire analysis was found for the GO term “T-cell activation” (GO: 0042110), which contained 92 genes (supplemental Table 5) relevant for the CLL/T-cell cross talk. Genes upregulated in Tcl-1 tg IRF4 ΔB/ΔB mice (Figure 2B) were mainly associated with intracellular signaling like Zap70,48 Ripk3, Mtor, and Myb, whereas the downregulated gene cluster (Figure 2B) mainly contained genes involved in antigen processing and presentation (MHC1/2, CD74, Pycard, Fcer1g, March1) and B-cell surface markers involved in either T-cell activation (CD80/CD86) or T-cell exhaustion (PD-L1/PD-L2). For RNA-Seq validation, we performed real-time polymerase chain reaction (PCR) of selected genes and verified downregulation of MHC1 genes (Figure 2C), MHC2 genes (Figure 2D), the central regulator of MHC2 expression CIITA (Figure 2E), and CD80/CD86 (Figure 2F). In summary, IRF4 deletion was associated with enhanced expression of cell cycle–associated genes, as already reported for murine IRF4 germline-deficient CLL models49,50 and downregulation of genes involved in T-cell activation, potentially contributing to enhanced CLL tumor immune evasion, which has not been studied so far. Thus, we further characterized the surface expression of critical molecules involved in the CLL/T-cell cross talk in IRF4-proficient and IRF4-deficient Tcl-1 tg mice on the B- and T-cell side.
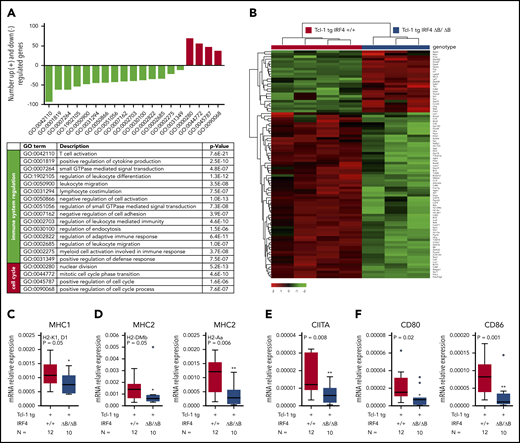
RNA-Seq and real-time PCR validation in Tcl-1 tg IRF4 WT and deficient CLL cells. (A) Number of significantly downregulated (negative y-axis, green bars) and upregulated genes (positive y-axis, red bars) corresponding to enriched GO terms (x-axis) in Tcl-1 tg IRF4 ΔB/ΔB (N = 3) as compared with Tcl-1 tg IRF4+/+ mice (N = 4). GO term descriptions and P values are indicated in the table. (B) Heatmap of differentially regulated genes within the GO term 0042110 T-cell activation. The counts per million were used for heatmap generation; unsupervised clustering (Euclidean) was used. (C) Validation of RNA-Seq data by real-time PCR in purified CLL cells derived from the spleen of Tcl-1 tg IRF4+/+ (red) and Tcl-1 tg IRF4 ΔB/ΔB (blue) mice. PCR was performed with TaqMan primers for the MHC1 genes H2-K1 and D1, (D) the MHC2 genes H2-Dmb and H2-Aa, (E) the MHC2 transactivator CIITA, and (F) the costimulatory molecules CD80 and CD86. Real-time PCR data were normalized to 18S and the mRNA relative expression ratio calculated according to the delta CT method.
RNA-Seq and real-time PCR validation in Tcl-1 tg IRF4 WT and deficient CLL cells. (A) Number of significantly downregulated (negative y-axis, green bars) and upregulated genes (positive y-axis, red bars) corresponding to enriched GO terms (x-axis) in Tcl-1 tg IRF4 ΔB/ΔB (N = 3) as compared with Tcl-1 tg IRF4+/+ mice (N = 4). GO term descriptions and P values are indicated in the table. (B) Heatmap of differentially regulated genes within the GO term 0042110 T-cell activation. The counts per million were used for heatmap generation; unsupervised clustering (Euclidean) was used. (C) Validation of RNA-Seq data by real-time PCR in purified CLL cells derived from the spleen of Tcl-1 tg IRF4+/+ (red) and Tcl-1 tg IRF4 ΔB/ΔB (blue) mice. PCR was performed with TaqMan primers for the MHC1 genes H2-K1 and D1, (D) the MHC2 genes H2-Dmb and H2-Aa, (E) the MHC2 transactivator CIITA, and (F) the costimulatory molecules CD80 and CD86. Real-time PCR data were normalized to 18S and the mRNA relative expression ratio calculated according to the delta CT method.
IRF4 deleted Tcl-1 tg tumors downregulate surface expression of molecules involved in CLL/T-cell interactions on the CLL and the T-cell side
CLL/T-cell interactions during antigen recognition, which is assumed to be crucial in CLL development and maintenance, consist of a number of interactions including antigen presentation on MHC 1/2 molecules and a complex surface expression pattern transmitting positive signals (CD80/CD86-CD28), negative signals (CD80/CD86-CTLA4), and exhaustion signals (PD-L1-PD-1).6,51 The percentage of MHC1+ CLL cells (Figure 3A) and the MHC2 MFIR was significantly downregulated in Tcl-1 tg IRF4 ΔB/ΔB mice in the spleen and lymph nodes (Figure 3B), but not in healthy C57/BL6 IRF4+/+ or ΔB/ΔB littermates (supplemental Figure 3A). The expression of CD80 (Figure 3C) and CD86 (Figure 3D) was severely decreased in Tcl-1 tg IRF4-deficient as compared with IRF4-proficient mice but not significantly different from tumor-free C57/BL6 littermates (supplemental Figure 3B). This suggests failed upregulation instead of active downregulation of CD80/CD86 during CLL tumor development. CD80/CD86 may interact with either CD28 or CTLA4 on the T-cell side, leading to stimulatory or inhibitory signals, respectively. CTLA4 was expressed very close to the detection limit, whereas CD28 was significantly decreased in CD8+ T cells derived from the spleen of IRF4-deficient Tcl-1 tg mice (Figure 3E) and in CD4+ as well as CD8+ T cells derived from the lymph nodes (Figure 3F; supplemental Figure 3C). Next, we evaluated the exhaustion markers PD-L1 and PD-1, which were reported to contribute to the T-cell exhaustion phenotype characteristic for CLL.12,16 In fact, PD-L1 and PD-1 were upregulated in the spleen (Figure 3G) and the lymph nodes (Figure 3H) of Tcl-1 tg IRF4+/+ mice compared with healthy C57/BL6 littermates. By contrast, PD-L1 upregulation was not observed on CLL cells derived from the spleen of Tcl-1 tg IRF4 ΔB/ΔB mice and the percentage of PD-1 positive T cells dropped to percentages typical for nonleukemic C57/BL6 mice (Figure 3G-H; supplemental Figure 3D). Thus, lack of IRF4 contributed to tumor immune evasion by downregulation of antigen presentation and costimulation on the B-cell side and eliminated the need of T-cell exhaustion. Therefore, we aimed to further characterize the effect of CLL-specific IRF4 deletion on the T-cell compartment.
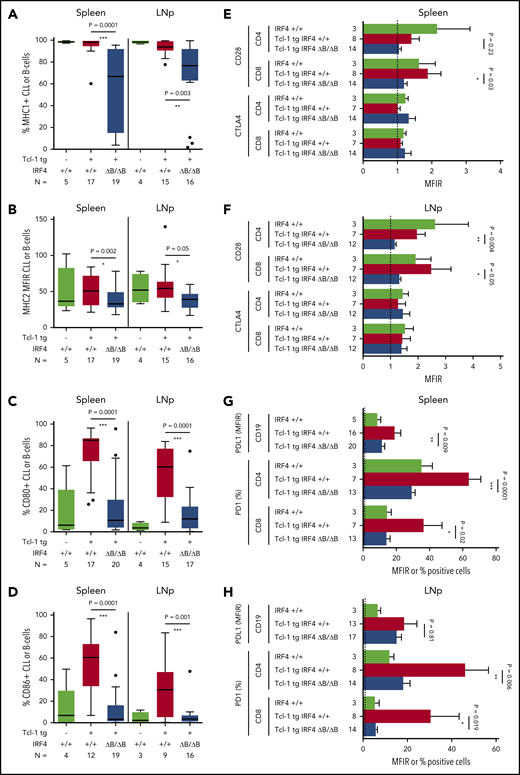
Surface marker expression in Tcl-1 tg IRF4+/+ and Tcl-1 tg IRF4 ΔB/ΔB mice. (A) MHC1, (B) MHC2, (C) CD80, and (D) CD86 surface expression was measured by flow cytometry in healthy C57/BL6 WT littermates (green), Tcl-1 tg IRF4+/+ (red), and Tcl-1 tg IRF4 ΔB/ΔB (blue) CD19+ B cells or CD19+ CD5+ CLL cells derived from either the spleen or the LNp. All measurements were performed with a proper isotype control to define negative cell populations or to calculate the MFIR. (E) The surface expression of CTLA4 and CD28 was measured in CD8 or CD4+ T cells using flow cytometry in cells derived from the spleen or (F) the lymph nodes. MFIR were calculated using an isotype control. Analyzed mouse numbers are depicted on the y-axis. (G) Flow cytometry of PD-L1 MFIR in CLL cells and percent PD-1+ cells in either CD4+ or CD8+ T cells. Genotypes and mouse numbers are depicted on the y-axis. All measurements were isotype controlled and were performed in cells derived from either the spleen or (H) the lymph nodes. MFIR, mean fluorescence intensity ratio.
Surface marker expression in Tcl-1 tg IRF4+/+ and Tcl-1 tg IRF4 ΔB/ΔB mice. (A) MHC1, (B) MHC2, (C) CD80, and (D) CD86 surface expression was measured by flow cytometry in healthy C57/BL6 WT littermates (green), Tcl-1 tg IRF4+/+ (red), and Tcl-1 tg IRF4 ΔB/ΔB (blue) CD19+ B cells or CD19+ CD5+ CLL cells derived from either the spleen or the LNp. All measurements were performed with a proper isotype control to define negative cell populations or to calculate the MFIR. (E) The surface expression of CTLA4 and CD28 was measured in CD8 or CD4+ T cells using flow cytometry in cells derived from the spleen or (F) the lymph nodes. MFIR were calculated using an isotype control. Analyzed mouse numbers are depicted on the y-axis. (G) Flow cytometry of PD-L1 MFIR in CLL cells and percent PD-1+ cells in either CD4+ or CD8+ T cells. Genotypes and mouse numbers are depicted on the y-axis. All measurements were isotype controlled and were performed in cells derived from either the spleen or (H) the lymph nodes. MFIR, mean fluorescence intensity ratio.
B cell–specific IRF4 deletion alters the CLL typical T-cell subset skewing
Tcl-1 tg mice show an antigen-experienced effector memory, but exhausted T-cell phenotype.12,15 In IRF4-deficient Tcl-1 tg mice, PD-1 and PD-L1, however, were not upregulated, suggesting that T-cell differentiation was altered. Therefore, we characterized naïve, central memory (TCM) and effector memory (TEM) T cells based on their CD44 and CD62L expression in CD4+ and CD8+ T cells using single-cell mass cytometry and found a relative increase of naïve T cells over antigen-experienced TEM/TCM T cells in Tcl-1 tg IRF4 ΔB/ΔB compared with Tcl-1 tg IRF+/+ mice (Figure 4A). These results were validated by classical flow cytometry (supplemental Figure 4A) in CD4+ T cells (Figure 4B-C) and CD8+ T cells (Figure 4D-E). The distribution of naïve vs antigen-experienced TEM/TCM T cells in IRF4-deficient Tcl-1 tg mice was thus similar to nonleukemic C57/BL6 littermates with a dominantly naïve CD4+ and CD8+ T-cell system (supplemental Figure 4B) and low numbers of antigen-experienced TEM/TCM T cells (supplemental Figure 4C). Next, we analyzed exhaustion markers in different T-cell subsets using single-cell mass cytometry (Figure 4F) and classical flow cytometry. CD223 (Lag-3) was expressed mainly in antigen-experienced T-cell subsets, whereas CD244 and CD160 expression was very low throughout all T-cell subsets (Figure 3G). No significant differences were observed between the genotypes (supplemental Figure 4D-F). PD-1 expression was mainly observed in CD4+ and CD8+ TEM/TCM T cells derived from IRF4-proficient and to a lesser extent from those derived from IRF4-deficient Tcl-1 tg mice, whereas naïve T cells were almost negative for PD-1 in both genotypes (Figure 4H). Using flow cytometry, we observed a significant downregulation of PD-1 in CD4+ TEM T cells derived from IRF4-deficient Tcl-1 tg mice, but not in CD4+ TCM T cells and CD8+ TEM/TCM T cells (Figure 4H). This suggests that the majority of T cells derived from Tcl-1 tg IRF4 ΔB/ΔB mice did not require T-cell exhaustion because of a disturbed CLL/T-cell cross talk. In addition, the decreased expression of CD28 in Tcl-1 tg IRF4 ΔB/ΔB mice (Figure 3E-F) was caused by an overrepresentation of naïve T cells over TEM/TCM T cells and not by active downregulation (supplemental Figure 4G-I). To draw a more direct connection between B cell–specific IRF4 deficiency and T-cell phenotypes, we performed in vitro cocultures of healthy C57/BL6 T cells with either Tcl-1 tg IRF4+/+ or Tcl-1 tg IRF4 ΔB/ΔB tumors for 72 hours. Although the percentage and absolute numbers of naïve CD4+ T cells did not change in cocultures with IRF4-deficient tumors, a decrease of naïve T cells (Figure 4I; supplemental Figure 4J) accompanied by an increase in antigen-experienced TEM/TCM CD4+ T cells (supplemental Figure 4K) was observed in cocultures with Tcl-1 tg IRF4+/+ tumors. In CD8+ T cells, a similar skewing from naïve to antigen-experienced T cells took place in presence of Tcl-1 tg IRF4-proficient but not in presence of IRF4-deficient tumors (Figure 4J; supplemental Figure 4L-M). Hence, the absence of IRF4 expression in CLL cells rendered CLL cells “less visible” to T cells, which partially prevented the differentiation of naïve T cells into antigen or tumor experienced T-cell subsets and eliminated the need of T-cell exhaustion.
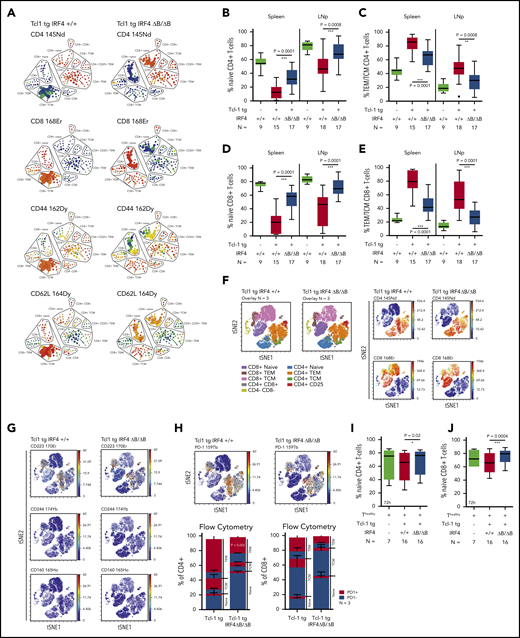
Naïve and antigen-experienced T-cell subsets in Tcl-1 tg IRF4-proficient and IRF4-deficient mice and in vitro cocultures. (A) SPADE (spanning-tree progression of density normalized events) analysis from single-cell mass cytometry data (Helios, CyTOF) showing the splenic T-cell compartment of leukemic Tcl-1 tg IRF4+/+ or Tcl-1 tg ΔB/ΔB mice. The color code represents staining intensities ranging from blue (no expression) to red (high expression). (B) Naïve and (C) antigen-experienced CD4+ T cells derived from the spleen or the lymph nodes of Tcl-1 tg IRF4+/+ or Tcl-1 tg ΔB/ΔB leukemic mice measured by flow cytometry. (D) Naïve and (E) antigen-experienced CD8+ T cells derived from the spleen or the lymph nodes of Tcl-1 tg IRF4+/+ or Tcl-1 tg ΔB/ΔB leukemic mice measured by flow cytometry. (F) Single-cell mass cytometry data showing the splenic T-cell compartment of leukemic Tcl-1 tg IRF4+/+ and Tcl-1 tg ΔB/ΔB mice. Color-coded viSNE overlay plots assign T-cell subsets to the TSNE1/TSNE2 coordinate system (left). viSNE intensity plots (blue = no expression to red = high expression) of CD4 and CD8 are shown on the right. (G) Expression of CD223 (Lag-3), CD224, CD160, and (H) PD-1 in the splenic T-cell compartment of leukemic Tcl-1 tg IRF4+/+ and Tcl-1 tg ΔB/ΔB mice. All viSNE plots are derived from the same experiment and show concatenated information of 3 mice per genotype. Data were validated using classical flow cytometry and results are shown as a stacking plot. PD-1+ cells are depicted in red, PD-1− cells in blue in naïve, TCM, and TEM CD4+ or CD8+ T cells. (I) Naïve CD4+ and (J) CD8+ T cells derived from healthy C57/BL6 IRF4+/+ mice measured 72 hours after coculture with Tcl-1 tg IRF4+/+ (red) or Tcl-1 tg IRF4 ΔB/ΔB (blue) tumors derived from the spleen of leukemic mice. Green box plots show control CD4+ or CD8+ T cells cultured in absence of tumor cells. TEM/TCM represents the total amount of both TEM and TCM cells. 145Nd, 168Er, 162Dy, 164Dy, 170Er, 174Yb, 165Ho, and 159Tb refer to rare metal conjugates of antibodies used in single-cell mass cytometry.
Naïve and antigen-experienced T-cell subsets in Tcl-1 tg IRF4-proficient and IRF4-deficient mice and in vitro cocultures. (A) SPADE (spanning-tree progression of density normalized events) analysis from single-cell mass cytometry data (Helios, CyTOF) showing the splenic T-cell compartment of leukemic Tcl-1 tg IRF4+/+ or Tcl-1 tg ΔB/ΔB mice. The color code represents staining intensities ranging from blue (no expression) to red (high expression). (B) Naïve and (C) antigen-experienced CD4+ T cells derived from the spleen or the lymph nodes of Tcl-1 tg IRF4+/+ or Tcl-1 tg ΔB/ΔB leukemic mice measured by flow cytometry. (D) Naïve and (E) antigen-experienced CD8+ T cells derived from the spleen or the lymph nodes of Tcl-1 tg IRF4+/+ or Tcl-1 tg ΔB/ΔB leukemic mice measured by flow cytometry. (F) Single-cell mass cytometry data showing the splenic T-cell compartment of leukemic Tcl-1 tg IRF4+/+ and Tcl-1 tg ΔB/ΔB mice. Color-coded viSNE overlay plots assign T-cell subsets to the TSNE1/TSNE2 coordinate system (left). viSNE intensity plots (blue = no expression to red = high expression) of CD4 and CD8 are shown on the right. (G) Expression of CD223 (Lag-3), CD224, CD160, and (H) PD-1 in the splenic T-cell compartment of leukemic Tcl-1 tg IRF4+/+ and Tcl-1 tg ΔB/ΔB mice. All viSNE plots are derived from the same experiment and show concatenated information of 3 mice per genotype. Data were validated using classical flow cytometry and results are shown as a stacking plot. PD-1+ cells are depicted in red, PD-1− cells in blue in naïve, TCM, and TEM CD4+ or CD8+ T cells. (I) Naïve CD4+ and (J) CD8+ T cells derived from healthy C57/BL6 IRF4+/+ mice measured 72 hours after coculture with Tcl-1 tg IRF4+/+ (red) or Tcl-1 tg IRF4 ΔB/ΔB (blue) tumors derived from the spleen of leukemic mice. Green box plots show control CD4+ or CD8+ T cells cultured in absence of tumor cells. TEM/TCM represents the total amount of both TEM and TCM cells. 145Nd, 168Er, 162Dy, 164Dy, 170Er, 174Yb, 165Ho, and 159Tb refer to rare metal conjugates of antibodies used in single-cell mass cytometry.
B cell–specific IRF4 deletion alters T-cell activation and IFN-γ secretion in Tcl-1 tg mice and enhances tumor cell engraftment in immunocompetent but not in immunodeficient recipients
In vivo, CLL T cells are activated by various signals from the microenvironment, leading to an antigen-experienced TEM/TCM T-cell phenotype in CD4+ and CD8+ T cells, which was absent in IRF4-deleted Tcl-1 tg mice. To further study the decrease in T-cell activation in relation to IRF4 deficiency in vitro, we used CD3/CD28-activated T cells derived from healthy C57/BL6 mice in coculture with CLL tumor cells derived from either Tcl-1 tg IRF4+/+ or Tcl-1 tg IRF4 ΔB/ΔB mice. T-cell activation defined by CD69 expression was significantly decreased in cocultures with Tcl-1 tg IRF4-deficient as compared with Tcl-1 tg IRF4+/+ tumors after 24 hours (Figure 5A) and a decreased skewing to memory T cells after 72 hours was observed (supplemental Figure 5A). This was also found in CD8+ T cells (Figure 5B; supplemental Figure 5B) and associated with decreased proliferation in both T-cell subsets (supplemental Figure 5C). Additionally, we observed decreased interferon-γ (IFN-γ) secretion (Figure 5C), which was more pronounced in CD8+ T cells (Figure 5D). Thus, the coincubation of healthy T cells with IRF4-deficient Tcl-1 tg tumors led to insufficient T-cell activation in vitro, suggesting a decreased antitumor recognition capacity in this setting. To verify this hypothesis in vivo, we transplanted spleen-derived tumor cells into immunocompetent C57/BL6 wild-type and immunodeficient NOD-SCID recipients. As shown in Figure 5E, IRF4-deficient Tcl-1 tg tumors engrafted earlier in immunocompetent recipients (42 days) as compared with IRF4-proficient tumors (92 days). In absence of an immunocompetent microenvironment, these differences vanished and overall survival was 26.5 and 33 days, respectively. Recipient T cells were skewed from naïve to antigen-experienced CD4+ and CD8+ T subsets in mice transplanted with Tcl-1 tg IRF4-proficient tumors but not in those transplanted with IRF4-deficient tumors (Figure 5F-G). Thus, IRF4 deficiency enhanced tumor immune evasion after transplantation leading to a survival benefit for transplanted CLL cells in immunocompetent recipients.
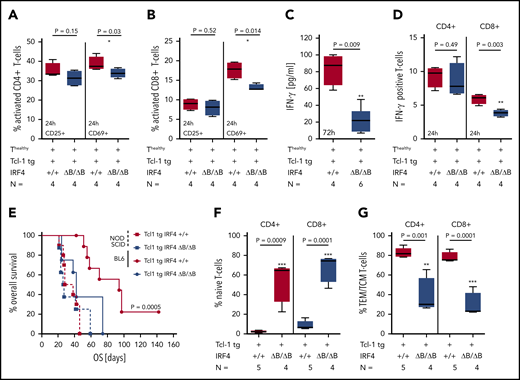
In vitro activation of healthy T cells and adoptive transfer. (A) CD4+ and (B) CD8+ T cells derived from healthy C57/BL6 IRF4+/+ mice were activated with CD3/CD28 beads and coincubated with CLL tumor cells derived from the spleen of either Tcl-1 tg IRF4+/+ or Tcl-1 tg ΔB/ΔB leukemic mice for 24 hours. The activation status of T cells was measured using CD25 and CD69 antibodies by flow cytometry. (C) IFN-γ secretion was measured 72 hours after CD3/CD28 bead activation in cell culture supernatants using enzyme-linked immunosorbent assay and (D) intracellular flow cytometry. (E) CLL tumor cells derived from the spleen of either Tcl-1 tg IRF4+/+ (red) or Tcl-1 tg ΔB/ΔB (blue) leukemic mice were transplanted into C57/BL6 WT (solid lines) or NOD-SCID recipients (dashed lines) using intraperitoneal injection. Overall survival of recipient mice is shown as Kaplan-Maier analysis. (F) Naïve and (G) antigen-experienced CD4+ and CD8+ T cells derived from the spleen of Tcl-1 tg IRF4+/+ or Tcl-1 tg ΔB/ΔB tumors transplanted in C57/BL6 recipients.
In vitro activation of healthy T cells and adoptive transfer. (A) CD4+ and (B) CD8+ T cells derived from healthy C57/BL6 IRF4+/+ mice were activated with CD3/CD28 beads and coincubated with CLL tumor cells derived from the spleen of either Tcl-1 tg IRF4+/+ or Tcl-1 tg ΔB/ΔB leukemic mice for 24 hours. The activation status of T cells was measured using CD25 and CD69 antibodies by flow cytometry. (C) IFN-γ secretion was measured 72 hours after CD3/CD28 bead activation in cell culture supernatants using enzyme-linked immunosorbent assay and (D) intracellular flow cytometry. (E) CLL tumor cells derived from the spleen of either Tcl-1 tg IRF4+/+ (red) or Tcl-1 tg ΔB/ΔB (blue) leukemic mice were transplanted into C57/BL6 WT (solid lines) or NOD-SCID recipients (dashed lines) using intraperitoneal injection. Overall survival of recipient mice is shown as Kaplan-Maier analysis. (F) Naïve and (G) antigen-experienced CD4+ and CD8+ T cells derived from the spleen of Tcl-1 tg IRF4+/+ or Tcl-1 tg ΔB/ΔB tumors transplanted in C57/BL6 recipients.
Decreased IRF4 expression in human CLL patients is related to inferior outcome, decreased T-cell skewing, and downregulation of genes involved in tumor immune evasion
Having demonstrated that IRF4 deficiency enhanced tumor immune evasion in murine CLL, caused by a downregulation of genes involved in antigen processing and T-cell activation, we next aimed to validate these results also in the human disease. We performed real-time PCR of purified CD19+ CLL or B cells derived from 98 chemo-naïve, unselected CLL patients and 9 healthy donors, respectively. IRF4 expression was significantly downregulated in CLL patients compared with healthy individuals and showed large interpatient variability (Figure 6A). Thus, we applied receiver operating characteristics (ROC) analysis using the time to first treatment (TTFT) as parameter separating IRF4low and IRF4high CLL patients. In Kaplan-Maier analysis (Figure 6B), IRF4low patients showed significantly decreased TTFT periods (51.3 month) compared with IRF4high CLL patients (79.4 months). No correlation of IRF4 expression to established CLL risk factors was observed (supplemental Figure 6A). In addition, we did not detect a different response to in vitro B-cell receptor stimulation between IRF4low and IRF4high CLL patients (supplemental Figure 6B). To independently validate our data, we analyzed the public domain GSE39671 dataset46 containing Affymetrix Gene chip data from 130 untreated CLL patients including TTFT data. The median TTFT was 29.8 months in IRF4low CLL patients and was not reached in IRF4high CLL patients (Figure 6C). The negative prognostic impact of decreased IRF4 expression could thus be validated in 2 independent CLL patient cohorts. Next, we determined differential gene expression from the GSE3967146 Affymetrix Gene chip dataset between IRF4low and IRF4high CLL patients. Of 92 differentially expressed genes found in the mouse system, 35 gene homologs (supplemental Table 6) also separated IRF4low and IRF4high CLL patients in unsupervised clustering (Figure 6D). Downregulated genes in IRF4low CLL patients included: CD86, CIITA, CD74, and HLA-DOA, which is in line with genes found in murine CLL. Next, we measured T-cell subsets in peripheral blood samples of CLL patients and found a correlation of low IRF4 expression to decreased percentages of antigen-experienced CD4+ (Figure 6E) and CD8+ T cells (Figure 6F). This correlation was found in IgVH-mutated and IgVH-unmutated CLL patients for the CD4+ compartment (supplemental Figure 6C) and in IgVH-unmutated CLL patients for the CD8+ T-cell compartment (supplemental Figure 6D). In addition, we detected a strong correlation of reduced IRF4 expression to reduced CD86 protein (Figure 6G) and CIITA (MHC2 class 2 transactivator) messenger RNA (mRNA) expression (Figure 6H), while HLA-B (supplemental Figure 6E) and HLA-DR (supplemental Figure 6F) mRNA was decreased only by trend in IRF4low as compared with IRF4high CLL patients. We thus speculated that differences in MHC gene expression in IRF4low vs IRF4high CLL patients might be more pronounced in active microenvironments, which prompted us to analyze the public domain dataset GSE21029,47 which contains Affymetrix Gene chip data from bone marrow (BM), lymph node (LN), or peripheral blood samples derived from primary CLL patients. We observed a downregulation of a variety of MHC genes in the BM (Figure 6I) of IRF4low CLL patients, whereas separation of MHC genes by IRF4 expression was less pronounced in LNs and the peripheral blood (supplemental Figure 6G). In addition, we performed IRF4 small interfering RNA knockdown in the MEC1 cell line, which was originally established from a prolymphocytic-transformed CLL patient52 and observed a significant downregulation of the MHC genes HLA-B and HLA-DR as well as of CD80 and CD86 (supplemental Figure 6H). Thus, we concluded that IRF4 was involved in the regulation of CD80 and CD86 expression in the human and murine disease, whereas MHC gene expression was regulated by IRF4 in a microenvironment-dependent manner, with the strongest association found in the BM. We also observed a connection of low IRF4 expression to inferior prognosis and failed T-cell skewing in human CLL, which was likely the result of a disturbed CLL/T-cell cross talk.
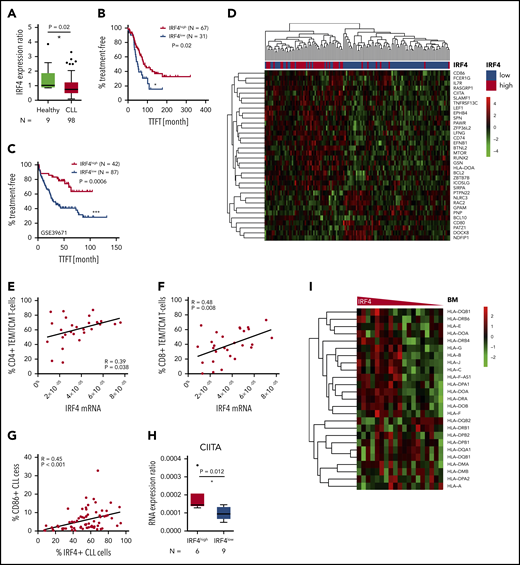
Relation of IRF4 expression to disease progression and the immune phenotype in human CLL patients. (A) Primary CLL cells derived from the blood of 98 chemonaive CLL patients and B cells derived from the blood of 9 healthy donors were purified and mRNA measured by real-time PCR. The mRNA expression ratio normalized to 18S and the mean expression in healthy donors is shown. (B) Kaplan-Maier analysis showing TTFT intervals for the IRF4low and IRF4high CLL patient group. The cutoff to discriminate between IRF4 groups was calculated by ROC analysis and Youden index calculation. (C) GSE39671 data46 were used for Kaplan-Maier analysis showing TTFT intervals in the IRF4low and IRF4high group. Group cutoffs were calculated using ROC analysis and Youden index calculation. (D) GSE39671 gene expression data46 were analyzed for differentially expressed genes (P < .05) in IRF4low and IRF4high CLL patients. Differentially expressed genes were filtered using the murine gene list GO: 0042210 and are depicted as heatmap using unsupervised clustering. (E) The percentage of CD4+ and (F) CD8+ naïve T cells and (G) CD86+ CLL cells was analyzed by flow cytometry and correlated to IRF4 expression measured by real-time PCR. (H) CIITA was measured by real-time PCR in IRF4low and IRF4high CLL patients. CIITA expression ratios are depicted as delta CT ratio, normalized to 18S. (I) Heatmaps of HLA genes extracted from Affymetrix Gene chip data derived from the public dataset GSE2102947 are shown for BM samples of CLL patients. IRF4 expression is shown on the x-axis using a declining expression gradient.
Relation of IRF4 expression to disease progression and the immune phenotype in human CLL patients. (A) Primary CLL cells derived from the blood of 98 chemonaive CLL patients and B cells derived from the blood of 9 healthy donors were purified and mRNA measured by real-time PCR. The mRNA expression ratio normalized to 18S and the mean expression in healthy donors is shown. (B) Kaplan-Maier analysis showing TTFT intervals for the IRF4low and IRF4high CLL patient group. The cutoff to discriminate between IRF4 groups was calculated by ROC analysis and Youden index calculation. (C) GSE39671 data46 were used for Kaplan-Maier analysis showing TTFT intervals in the IRF4low and IRF4high group. Group cutoffs were calculated using ROC analysis and Youden index calculation. (D) GSE39671 gene expression data46 were analyzed for differentially expressed genes (P < .05) in IRF4low and IRF4high CLL patients. Differentially expressed genes were filtered using the murine gene list GO: 0042210 and are depicted as heatmap using unsupervised clustering. (E) The percentage of CD4+ and (F) CD8+ naïve T cells and (G) CD86+ CLL cells was analyzed by flow cytometry and correlated to IRF4 expression measured by real-time PCR. (H) CIITA was measured by real-time PCR in IRF4low and IRF4high CLL patients. CIITA expression ratios are depicted as delta CT ratio, normalized to 18S. (I) Heatmaps of HLA genes extracted from Affymetrix Gene chip data derived from the public dataset GSE2102947 are shown for BM samples of CLL patients. IRF4 expression is shown on the x-axis using a declining expression gradient.
Discussion
Here we report a novel B cell–specific role of IRF4 in regulating the CLL/T-cell cross talk and demonstrate that lack of IRF4 in murine CLL contributes to enhanced CLL tumor immune evasion and a more aggressive type of disease with short treatment-free survival. Enhanced disease progression was also observed in human IRF4low CLL patients in 2 independent patient cohorts. Thus, IRF4 has tumor-suppressive functions in CLL, which is in contrast to other hematological malignancies, such as multiple myeloma34,53 and some subtypes of diffuse large B-cell lymphoma,35,54,55 but in line with data obtained in chronic myeloid leukemia, B-cell acute lymphoblastic leukemia, and acute myeloid leukemia,36-38 suggesting that the transcriptional program regulated by IRF4 is highly dependent on the B-cell maturation stage and transformation context. In CLL, which resembles a malignancy of mature B cells, the tumor-suppressive function of IRF4 was previously described in 2 mouse models: VH11 IRF4 knockout mice developed CLL spontaneously50 and in New Zealand Black mice an accelerated CLL disease development was observed.49 An essential caveat of these analyses was the use of germline-deleted IRF4 models, in which CLL develops within an IRF4-deficient microenvironment, including IRF4-deficient T cells, which showed impaired T-cell cytotoxicity and antitumor response.33 Moreover, CD4-specific IRF4 deletion was associated with immune defects and T-cell exhaustion,56 and CD8-specific IRF4 deletion contributed in the establishment of chronic infections57 because of abrogated effector T-cell differentiation.22 These data suggest that enhanced CLL progression in IRF4 germline models might be influenced by an IRF4-deficient and antitumor immune-compromised T-cell compartment, making it difficult to dissect the contribution of IRF4 in CLL cells. Therefore, we studied CLL development in Tcl-1 tg mice with CD19-restricted IRF4 deletion, allowing for analysis of the specific effect of IRF4 deletion in CLL tumor cells. At time of euthanasia, the tumor mass was similar in Tcl-1 tg IRF4+/+ and Tcl-1 tg IRF4 ΔB/ΔB mice, with a similar organ infiltration pattern and CLL typical clonal patterns, arguing for a consistent definition of mice that needed to be euthanized and against a changed overall behavior of the accelerated disease (Figure 1). We are therefore the first to demonstrate the specific effect of IRF4 deficiency in CLL cells and the CLL-driven T-cell shaping.
Using RNA-Seq, we analyzed the IRF4-driven transcriptional program in purified CLL cells and found an upregulation of genes involved in cell-cycle processes in Tcl-1 tg IRF4 ΔB/ΔB mice (Figure 2A). However, our data from transplant studies into immunodeficient recipients showed very similar growth kinetics between the genotypes, suggesting that the effects of cell-cycle dysregulation may be limited. In addition, however, we discovered a severe downregulation of genes involved in antigen processing and presentation and genes involved in the CLL/T-cell cross talk (Figure 2B). This immune regulatory function of IRF4 in relation to the microenvironment has not been described in CLL or other B-cell malignancies so far and could, unlike effects on cell-cycle regulation, only be sufficiently studied in a conditional model.
T cells are known to be critical in CLL disease development.5,6 We have previously shown that T cells are skewed from naïve to antigen-experienced subsets.10,15 In human CLL, the skewing of CD4+ T cells to antigen-experienced subsets was associated with short treatment-free survival in CLL patients with unmutated IgVH genes,10 likely caused by enhanced interleukin-4 and CD40L ligation, which increase CLL survival and proliferation, respectively.10,44 In contrast, we observed a relation of decreased IRF4 expression with decreased antigen-experienced T cells in IgVH-mutated/unmutated CLL on the CD4+ T-cell side and a very pronounced correlation in IgVH-unmutated CLL on the CD8+ T-cell side. Both studies thus demonstrate that T-cell support by CD4+ T cells and tumor immune evasion are likely distinct mechanisms that may contribute to increased CLL progression independently. Both studies also suggest an ongoing cross talk between CLL cells and T cells that is assumed to be antigen mediated and involves T-cell receptor/MHC binding, T-cell costimulation, and IFN-γ secretion.58 In fact, tumor-specific T cells were found in CLL patients, demonstrating that CLL-derived T cells retain cytotoxic antitumor activity.59 By contrast, Tcl-1 tg IRF4 ΔB/ΔB mice showed a downregulation of MHC molecules and decreased CD80/CD86 expression on CLL tumor cells (Figure 3A-D). In vitro, we observed decreased T-cell activation and reduced IFN-γ secretion and in vivo rapid growth of IRF-deficient tumors was observed in immunocompetent recipients (Figure 5A-E). In human CLL, we found a correlation of low IRF4 expression to decreased CD86 expression and alterations in the gene expression program (Figure 6), similar to the murine disease. Moreover, the percentage of differentiated antigen-experienced T cells was decreased in IRF4low CLL patients, indicating impaired CLL cell recognition by T cells and enhanced tumor immune evasion (Figure 6E-F). Defective CD4 T-cell priming was previously reported in a mouse model with IRF4 deficiency restricted to dendritic cells; this was due to decreased MHC class 2 antigen presentation and downregulation of genes involved in antigen processing,26 showing high overlap to downregulated genes observed in our study, including CIITA (Figure 6H; supplemental Table 3), a transactivator required for MHC2 expression25,26,60 and direct transcriptional IRF4 target.61
Thus, we suggest that the downregulation of genes relevant for the CLL/T-cell cross talk observed in Tcl-1 tg IRF4 ΔB/ΔB mice and IRF4low CLL patients, enhanced CLL tumor immune evasion, leading to decreased numbers of antigen-experienced, potentially tumor-specific T cells, and a more aggressive course of disease. Although some cytotoxic antitumor-specific T cells were found in human CLL patients,58,59 the majority of T cells is exhausted, which prevents an effective antitumor immune response.12,13,16 In line with previous results, we detected an upregulation of PD-L1 and PD-1 in Tcl-1 tg IRF4+/+ mice. By contrast, PD-L1 was downregulated in CLL tumor cells derived from Tcl-1 tg IRF4 ΔB/ΔB mice and PD-1 decreased (Figure 3G-H) in the CD4+ TEM compartment (Figure 4H). No differences in PD-1 expression were observed in the CD8+ T-cell compartment; however, in Tcl-1 tg IRF4 ΔB/ΔB mice, the majority of CD8+ (and CD4+) T cells showed a naïve phenotype. These naïve T cells are not tumor reactive and thus are not under the selection pressure to develop T-cell exhaustion.
We therefore conclude that at least 2 different mechanisms for tumor immune escape in CLL exist: (1) T-cell exhaustion, which is required to prevent an immune attack of CLL tumor cells by antigen-experienced T cells and (2) an immune privileged state afforded by IRF4 deficiency (or downregulation), which blocks CLL/T-cell interactions. In CLL patients, it is likely that both strategies are used at the same time, which is also relevant for tumor evolution after therapy.43,62,63 It has been reported that treatment of primary CLL patients with the BTK inhibitor Ibrutinib in vivo downregulated genes involved in immune response, including IRF4, PD-L1, and PD-1.63,64 Interestingly, patients that discontinued Ibrutinib showed poor overall survival afterwards,65 and it might be interesting to investigate a relation of this observation to the enhanced immune evasion capacity of IRF4low CLL patients described here. In addition, Ibrutinib is currently used in combination immunotherapy approaches with PD-1 inhibitors and CAR T cells.66 In the light of our findings, it may be important to investigate effects on T-cell quality and antigen presentation in such studies. Given that immunotherapies are the most promising option to cure CLL in the long term, we believe that molecular understanding of critical determinants of CLL tumor immune evasion is essential to devise strategies that combine modulation of such determinants, such as IRF4 expression with available immune treatment strategies to achieve this goal.
The publication costs of this article were defrayed in part by page charge payment. Therefore, and solely to indicate this fact, this article is hereby marked “advertisement” in accordance with 18 USC section 1734.
RNA sequencing datasets will be deposited to public repositories. Original data will be shared upon E-Mail request to the corresponding author.
The online version of this article contains a data supplement.
Acknowledgments
The authors thank the IIIrd Medical Department, University Hospital Salzburg, Austria, for providing patient material and patient data. The authors also thank Erika Nussbaumer and Nathalie Wacht for purification of PBMCs from peripheral blood of CLL patients and biobanking.
This study was supported by the SCRI-LIMCR GmbH, the Province of Salzburg (20102-P1509466-FPR01-2015 and 20102-P1601064-FPR01-2017 to R.G.), grants from the Austrian Science Fund FWF to D.A. (T671-B13) and A.E. (P26719-B19, I1299-B21, and I3282-B26 (FOR 2036), I2795-B28 (ERA-NET TRANSCAN-2 program JTC 2014; project FIRE-CLL), and the Paracelsus Medical Private University Salzburg (PMU E-18/28/147-EGA).
Authorship
Contribution: A.E. and D.A. designed the research and wrote the manuscript; D.A., Y.Q., N.M., U.D., M.S., and J.P.H. performed experimental work; D.A. and Y.Q. analyzed the data; A.E., D.A., Y.Q., N.M., M.S., N.Z., T.N.H., and R.G. contributed in critical discussion and data interpretation; and all authors read and agreed to the final version of the manuscript.
Conflict-of-interest disclosure: The authors declare no competing financial interests.
The current affiliation for T.N.H. is Department of Hematology, Oncology and Stem Cell Transplantation, Faculty of Medicine and Medical Center, University of Freiburg, Freiburg, Germany.
Correspondence: Alexander Egle, Department of Internal Medicine III, Müllner Hauptstraße 48, 5020 Salzburg; e-mail: a.egle@salk.at.

This feature is available to Subscribers Only
Sign In or Create an Account Close Modal