

Abstract
Thrombotic and hemorrhagic complications are prevalent in patients with essential thrombocythemia, polycythemia vera, and myelofibrosis. Given the impact on morbidity and mortality, reducing the risk of thrombosis and/or hemorrhage is a major therapeutic goal. Historically, patients have been risk stratified on the basis of traditional factors, such as advanced age and thrombosis history. However, multiple factors contribute to the thrombotic tendency, including gender, mutational profile, inflammatory stress, and abnormal cell adhesion. Management includes cardiovascular risk reduction and use of antiplatelet therapy, depending on myeloproliferative neoplasm subtype and mutational status. Anticoagulation is a mainstay of therapy for those with venous thrombosis, but practice patterns remain heterogeneous. Cytoreduction is indicated for higher-risk patients, but efficacy may depend on the involved vascular bed. Management of special situations, such as unusual site thrombosis, bleeding, the perioperative period, and pregnancy, are especially challenging. In this article, risk factors and treatment strategies for myeloproliferative neoplasm thrombosis and bleeding, including special situations, are reviewed. Insights gained from recent studies may lead to the development of a more precise risk classification and tailored therapy.
Clinical case
A 27-year-old woman presented with abdominal pain and distention after laparoscopic cholecystectomy. Six years earlier, leukocytosis (14 × 109/L) and thrombocytosis (615 × 109/L) had been noted. She had delivered her first child 4 months before her current presentation and now took oral contraceptives and smoked 3 cigarettes daily. Her evaluation revealed abdominal distention and tenderness together with a leukocyte count of 15.8 × 109/L, hemoglobin 15.6 g/dL (hematocrit [Hct], 49%), and platelets 614 × 109/L. Magnetic resonance imaging and a hepatic venogram confirmed ascites, hepatosplenomegaly, and chronic hepatic vein thrombosis. A liver biopsy noted histological findings consistent with venous outflow obstruction. Altogether, the findings suggested chronic Budd-Chiari syndrome (BCS). A bone marrow biopsy revealed normocellularity, a slight increase in megakaryocytes, and no increase in reticulin fibrosis. JAK2 V617F was identified.
Introduction
The classical myeloproliferative neoplasms (MPNs)—essential thrombocythemia (ET), polycythemia vera (PV), and myelofibrosis (MF)—are clonal stem cell disorders characterized by expansion of mature blood elements, extramedullary hematopoiesis, disease transformation, and a pronounced symptom burden. Thrombotic events are prevalent across MPN subtypes, especially near diagnosis; a recent meta-analysis, including 13 436 patients (49% ET, 35% PV, 14% MF), identified a pooled prevalence of overall thrombosis in 28.6%, 20.7%, and 9.5% of newly diagnosed patients with PV, ET, and MF, respectively.1 Consistent with prior observations, arterial thrombosis (pooled prevalence, 16.2%; cerebrovascular, 7.4%; transient ischemic attack, 3.5%; coronary heart disease, 6.1%) was more common than venous thrombosis (pooled prevalence, 6.2%; deep vein thrombosis, 3.4%; splanchnic vein thrombosis [SVT], 1.4%; pulmonary embolism, 0.9%; cerebral venous thrombosis [CVT], 0.7%).1 A recent population-based study demonstrated the magnitude of thrombosis risk: Among 9429 patients with MPN, thrombosis hazard ratios (HRs) were increased around the time of diagnosis, and also at later time points (HRs, 4.0, 2.4, and 1.8 at 3 months, 1 year, and 5 years after diagnosis, respectively), compared with age- and gender-matched control participants.2 The absolute number of arterial events was higher, but the magnitude of risk was lower, than in venous thrombosis (arterial HRs, 3.0, 2.0, and 1.5 vs venous HRs, 9.7, 4.7, and 3.2, respectively, at the same time points above). Confirming the influence of traditional risk factors, thrombosis risk was increased in those over 60 years of age and in those with prior thrombosis, and it was highest in patients with both of these risk factors.2 Because of its prevalence and impact on morbidity and mortality, reducing thrombosis risk is a major goal of therapy.
Though less prevalent than thrombosis, preventing or managing bleeding is equally important. In the aforementioned meta-analysis, the pooled prevalence of bleeding complications was 6.2%.1 In this study, as in others, MPN subtype affects bleeding risk; the highest prevalence was found in MF (8.9%), followed by ET (7.3%) and PV (6.9%).1,3,4 Common sites of bleeding include mucocutaneous, gastrointestinal, epistaxis, and postoperative.1
Effective management requires an understanding of the multiple factors that contribute to MPN-associated thrombosis and bleeding, which are reviewed in this article. Furthermore, we discuss management strategies with attention to special situations, including unusual site thrombosis, bleeding, the perioperative period, and pregnancy.
Thrombosis risk factors
Traditional risk factors
Advanced age and past history of thrombosis are consistently identified risk factors. The former was highlighted in a meta-analysis of 3236 patients treated with hydroxyurea (HU) in which thrombosis rates were 1.9%, 3.6%, and 6.8% at median ages of 60, 70, and 80, respectively.5 Statistical associations between cardiovascular (CV) risk factors and MPN thrombosis have been inconsistent in ET.6-9 Although CV factors are not included in PV risk classification, low-risk patients with PV with hypertension had a thrombosis-free survival of 34% compared with 66% of those without hypertension (P = .025).10 The retrospective nature and lack of distinction between arterial and venous thrombosis in most studies may dilute an intuitive impact of CV risk factors.8 In a prospective study of 258 patients with MPN, CV risk factors and hyperlipidemia were associated with arterial (but not venous) thrombosis in the entire cohort.11 However, CV risk factors for arterial thrombosis differed depending on MPN subtype: thrombosis risk factors included diabetes mellitus in PV, hypertension/hyperlipidemia in ET, and hyperlipidemia and at least one other CV risk factor in MF.
Blood cell counts
Erythrocytosis associates with CV outcomes in PV. In randomized study of 365 JAK2 V617F-positive patients with PV, those who achieved a Hct <45% using phlebotomy and/or cytoreduction had significantly lower rates of CV death and major thrombosis than those with an Hct of 45% to 50% (HR, 3.9; P = .007).12 The latter group also had higher leukocyte counts, and thrombosis risk was statistically increased when the leukocyte count was >11 × 109/L in a post hoc analysis.13 A statistically significant, nearly linear association between thrombosis and leukocyte count was also observed in a prospective study of ET (21 887 blood counts analyzed), though a particular leukocyte threshold beyond which risk increased was not identified.14 A recent meta-analysis including >30 000 patients with ET and PV also reported an association between leukocytosis and thrombosis (RR, 1.59; 95% confidence interval [CI], 1.4-1.8), mainly in ET (relative risk [RR], 1.65) and arterial thrombosis (RR, 1.45) subgroups.15 In MF thrombosis, leukocytosis was of borderline significance (P = .06), but thrombosis risk increased with leukocytes >15 × 109/L in the presence of JAK2 V617F.16 In prefibrotic/early MF, leukocytosis (>11.2 × 109/L) was associated with thrombosis risk.17 Thrombocytosis, however, has not strongly correlated with thrombosis risk.14
Mutational profile
Thrombosis risk differs by mutational status.9 Adjusted for age, CALR-ET had a lower 15-year cumulative incidence of thrombosis (10.5% vs 25.1%; P = .001) than JAK2 V617F-positive ET in a retrospective comparison.18 Similarly, another study found the 10-year cumulative incidence of thrombosis was lowest in those with CALR-ET and triple-negative ET (5.1% and 8.2%, respectively), compared with JAK2 and MPL-mutant ET (14.5% and 19.5%, respectively; P = .008).19 In a retrospective study of 617 patients with MF, the 10-year cumulative incidence of thrombosis was lower for CALR (both types 1 and 2) than for JAK2 V617F-mutant MF (13.6% vs 18.3%; P = .021) after adjustment for age.20 Increased JAK2 V617F allele burden has correlated with thrombosis risk in PV.21 Furthermore, JAK2 V617F allele burden may impact thrombosis type: values >25% were associated with arterial thrombosis (P = .055), and values >90% were associated with venous thrombosis (P = .036).11 Less is known about the impact of additional mutations on thrombosis risk. Among 316 patients (133 PV, 183 ET), TET2 mutations were associated with thrombosis risk in ET (P = .01), independent of age and driver mutational status.22 However, this association was not observed in PV or in an external cohort of 174 Italian patients.22
Inflammation
Because MPNs are characterized by chronic inflammation that may accelerate atherosclerosis, candidate inflammatory biomarkers, including pentraxin 3 (PTX3) and high-sensitivity C-reactive protein (hs-CRP), have been studied.9 In a cross-sectional study of 477 patients (305 ET, 172 PV), JAK2 V617F homozygosity was associated with high PTX3 levels; yet, thrombosis rates were lower at the highest PTX3 levels.23 PTX3 therefore may have a protective role, as supported by preclinical studies suggesting that PTX3 limits P-selectin–associated inflammation.23 On the contrary, those with hs-CRP levels above the 50th percentile had an increased risk of thrombosis (odds ratio [OR], 2.57; 95% CI, 1.39-4.75), disease progression (OR, 2.7), and death (OR, 3.93).23 Results suggest that MPN inflammation may impact both vascular risk and disease progression.
Abnormal cell adhesion
Novel mechanisms also contribute to the prothrombotic state (Figure 1).24 A proadhesive phenotype, mediated by increased P-selectin, was demonstrated in an in vitro and murine model expressing JAK2 V617F mutation in endothelial cells. This model also had increased spontaneous and inflammation-associated thrombus formation.25 Furthermore, a P-selectin–blocking antibody and HU reduced thrombus formation, leukocyte rolling and adhesion, endothelial cell release, and expression of P-selectin.25 JAK2 V617F endothelial lineage cells (redirected from induced pluripotent stem cells) were also prone to leukocyte adhesion, also mediated through increased P-selectin levels and a proinflammatory phenotype.26 These studies raise the possibilities of using P-selectin as a biomarker and anti–P-selectin antibodies (eg, crizanlizumab) as a therapy in MPN.24,25,27 Abnormal leukocyte adhesion may also be mediated by the activation of leukocyte integrins, as demonstrated by JAK2 V617F–induced activation of β1- and β2-integrins in a murine model.28 Here, neutralizing antibodies against integrins suppressed venous thrombus formation.28
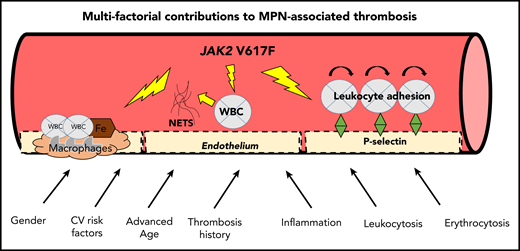
Multifactorial contributions to MPN-associated thrombosis. ASA, acetylsalicylic acid; WBC, white blood cells. Figure concept adapted in part from Setiadi et al.87
Multifactorial contributions to MPN-associated thrombosis. ASA, acetylsalicylic acid; WBC, white blood cells. Figure concept adapted in part from Setiadi et al.87
Another novel mechanism includes the formation of neutrophil extracellular traps (NETs). Wolach et al quantified increased NET formation in neutrophils from patients with MPN, compared with patients with myelodysplastic syndrome and age-matched control subjects.29 Furthermore, isolated neutrophils with increased NET formation and increased expression of PAD4, required for NET formation, were found in a JAK2 V617F heterozygous murine model with spontaneous pulmonary thrombosis. Treatment with ruxolitinib decreased NET formation in vitro and reduced thrombosis in the murine model.29 Interestingly, a higher rate of major venous thrombosis in those with JAK2 V617F clonal hematopoiesis of indeterminate potential (CHIP), compared with those without JAK2 V617F CHIP (P = .0003) or other CHIP mutations (P = .02), was also reported in this study. The authors hypothesized that this observation could be due to the presence of clonal neutrophils prone to NET formation.29
Finally, JAK2 V617F alters the lipid core of atherosclerotic plaque. Compared with JAK2–wild-type mice, hypercholesterolemic JAK2-mutant mice had earlier development of atherosclerosis, with increased neutrophil infiltration and macrophages and larger necrotic cores.30 These characteristics increase plaque instability and suggest that optimal control of lipids may be important.
Treatment considerations
Prevention of incident and/or recurrent thrombosis is a major goal of therapy (Figures 2 and 3). Management of CV risk factors is important, regardless of inclusion in risk classifications. In PV, evidence supports Hct control12 and low-dose aspirin, which offers a 60% risk reduction of adverse outcomes.31 Evidence is less convincing for universal aspirin use in ET. In a systematic review of 24 studies (none randomized) including 6153 patients, the certainty of evidence was rated low or very low for RRs of thrombosis, any bleeding, or major bleeding.32 In low-risk CALR-ET, compared with observation, antiplatelet therapy was associated with an increased risk of bleeding (12.9 vs 1.8 episodes per 1000 patient-years; P = .03), without a reduction in thrombosis risk.33 When used, the optimal frequency of aspirin has been questioned, and a study comparing various aspirin regimens and their impact on platelet thromboxane inhibition and clinical outcomes is ongoing.34 There are no clinical trials supporting use of alternative antiplatelet therapies.
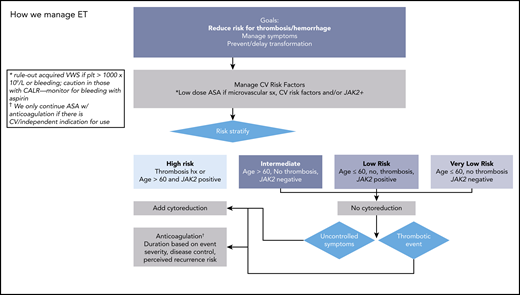
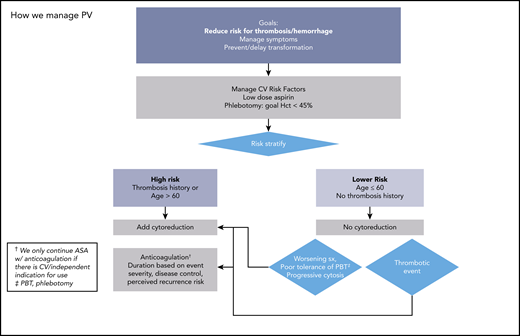
How we manage PV. ASA, acetylsalicylic acid. Adapted with permission from Stein and Gerds.88
How we manage PV. ASA, acetylsalicylic acid. Adapted with permission from Stein and Gerds.88
Cytoreduction is indicated in high-risk patients; evidence does not yet support its use in lower-risk patients.35 A randomized study comparing HU and aspirin with aspirin alone in 382 patients with ET, aged 40-59, and without high-risk features or extreme thrombocytosis found a low vascular event rate (0.93 per 100 patient-years) and no difference in a composite endpoint including time to arterial/venous thrombosis, serious bleeding, or vascular death (HR, 0.98; P = 1.0).36 In high-risk patients with ET and PV, HU is a typical first choice based on randomized data in ET and extrapolation in PV.35,37 Risk reduction with HU may depend on the involved vascular bed; in a study of 1500 patients, HU (as well as aspirin and vitamin K antagonists [VKAs]) reduced risk of recurrent arterial but not venous events.38 Pegylated interferon (IFN) is also a first-line cytoreductive agent, and although phase III studies with a novel IFN, ropeginterferon, vs HU in PV have been completed, detailed thrombosis event rates have not been published yet. Guidelines (National Comprehensive Cancer Network [NCCN]39 but not European LeukemiaNet40 ) consider anagrelide (ANA) as another front-line option, based on randomized data of 259 untreated high-risk patients with ET that demonstrated noninferiority when compared with HU for thrombotic or bleeding events.41 Ruxolitinib is a second-line option in PV, and 5-year follow-up data revealed lower rates of thromboembolic events in ruxolitinib randomized (1.2 per 100 patient-years) and crossover patients (2.7 per 100 patient-years) compared with best available therapy (8.2 per 100 patient-years; no P value reported).42 However, the thromboembolic event rate was not considered as a primary endpoint, so conclusions cannot be drawn. In a retrospective study of 1500 patients, in which limitations in design and sample size were noted, other cytoreductives (ANA, busulfan, IFN, pipobroman, and ruxolitinib) did not demonstrate antithrombotic efficacy.38 It should be noted that complete hematological response is not a surrogate for reduction in thrombosis risk in either patients with ET treated with HU43 or ANA44 or patients with PV treated with HU.45
Anticoagulation management for thrombosis remains heterogeneous.9 When 73 hematologists, including 58% MPN experts, were presented 5 thrombotic scenarios, lack of management consensus was evidenced by variation in antithrombotic use in all vignettes with ET/PV thrombosis.46 Studies reflect this heterogeneity. In one study, among 526 patients, 99 MPN-associated venous thromboembolisms (VTEs) were identified in 78 patients (7 untreated); VKAs were most commonly used (56%), followed by low-molecular-weight heparins (LMWHs) (24%) and direct oral anticoagulants (DOACs) (20%).47 Patients were anticoagulated for a median of 12 months; in about half (36 patients), the duration was 6 months. Stopping anticoagulation after 6 months significantly increased the risk of recurrence (36% vs 8.6%; P = .01) at a median of 10 months from discontinuation.47 Only limited studies have included patients treated with DOACs,38,47,48 and, on the basis of absence of high-quality data, NCCN guidelines do not advise on a specific anticoagulant choice.49 Duration of anticoagulation is based on event severity, degree of disease control, and likelihood for recurrence after discontinuation of anticoagulation.49
The treatment considerations described above largely apply to patients with ET and PV; there are no predictive models to estimate thrombosis risk in MF. However, a recent pragmatic treatment algorithm50 advises aspirin in patients with prefibrotic MF with prior arterial thrombosis (anticoagulation instead if prior venous thrombosis), advanced age, CV risk, JAK2 V617F, leukocytosis, or microvascular symptoms. HU is advised in those with thrombocytosis or leukocytosis, though specific thresholds are not included.50
Special situations
Unusual site thrombosis
Management of special situations poses a unique challenge (Figure 4). Workup for MPN is low yield in patients with CVT lacking myeloproliferation; of 706 patients with CVT, only 3.8% were diagnosed with MPN, and CVT was identified in only 0.4% of a cohort of 2143 patients with established MPN.51 To the contrary, MPN prevalence is higher among patients with SVT.52 A meta-analysis reported a prevalence of MPNs in 40.9% of patients with BCS (PV, 52.9%; ET, 24.6%), including 17.1% who initially lacked typical hematological abnormalities that became evident 0.7 to 7 years later.53 In those with portal vein thrombosis (PVT), the MPN prevalence was 31.5% (PV, 27.5%; ET, 26.2%; solitary JAK2-positive MPN, 24%), including 15.4% who initially lacked typical hematologic abnormalities but in whom abnormalities appeared 1 to 10 years later.53 That SVT is often the presenting feature of MPN was also confirmed by a population-based study, which reported an HR of SVT of 81.1 within 3 months of MPN diagnosis compared with those without MPN.2
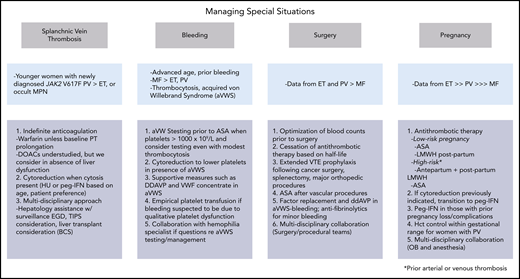
Managing special situations. DDAVP, desmopressin; EGD, esophagogastroduodenoscopy; OB, obstetrics; VWF, von Willebrand factor.
Managing special situations. DDAVP, desmopressin; EGD, esophagogastroduodenoscopy; OB, obstetrics; VWF, von Willebrand factor.
MPN-SVT typically presents in newly diagnosed, younger women.52 When patients with PV aged <45 years at diagnosis were compared with those aged >65 years at diagnosis, overall thrombosis prevalence was similar between the groups, despite the younger cohort having lower leukocytes and JAK2 V617F allele burdens.54 However, venous events were more common in younger patients, especially those with SVT (13% vs 2%; P = .006). Among the younger patients, 76% were women, and SVT was typically the first manifestation of their MPN.54 Younger age (47 vs 56.5 years; P = .003), female predominance in PV (67% vs 37%; P = .02), and lower JAK2 V617F allele burden (median, 5% vs 36.3%; P = .019) were also found in patients with MPN-SVT (n = 52) compared with patients with MPN without SVT (n = 134).55
Most patients with MPN-SVT have JAK2 V617F rather than other driving mutations.53 No patient in the SVT meta-analysis harbored a JAK2 exon 12 mutation, and only 3 of 305 tested patients carried MPL mutations.53 In another systematic review, the pooled proportion of CALR mutations was 1.2% in all patients with SVT (1.4% in BCS, 1.6% in PVT).56 Rather than routinely testing for CALR in patients with SVT, restricting testing to JAK2 V617F–negative patients, spleen size ≥16 cm, and platelets >200 × 109/L avoids unnecessary testing.57
Because JAK2 V617F is commonly identified in SVT, it is likely involved in pathogenesis.52 An early study identified JAK2 V617F in endothelial cells from patients with PV with BCS, suggesting that abnormal adhesion may play a prominent role.58 Haplotype may also impact risk. A pooled analysis of 26 studies identified an association between the JAK2 46/1 haplotype and an increased risk of both MPN and SVT,59 and another study suggested that an association between the 46/1 haplotype and JAK2 V617F acquisition in women with SVT could explain the gender difference in SVT.60
A recent algorithm advises indefinite anticoagulation with VKA for patients with SVT.61 However, despite a high proportion of VKA use (85%), one study of 181 patients with MPN-SVT found a recurrence rate of 3.9 per 100 patient-years, which was similar to patients with SVT who either discontinued or never received VKAs (5.4 per 100 patient-years; P = .41).62 Patients with BCS were at particular risk for recurrence, as were those with prior thrombosis history, splenomegaly, and leukocytosis (>14 × 109/L).62 In another small study of MPN-SVT, treatment strategy, including systemic anticoagulation, did not influence recurrence.63 Whether LMWH or DOACs would be more effective than VKA is unknown. In a registry study of DOAC use in MPNs, no patient had SVT.48 Importantly, though indefinite anticoagulation is indicated, patients with MPN-SVT are at higher risk for bleeding; in a prior series, major bleeding was observed at a rate of 2.1 per 100 patient-years.62
Adding cytoreduction for MPN-SVT is also a standard recommendation. However, among 1500 patients with MPN, HU did not impact the rate of recurrence in 218 patients with SVT.38 Because reduction in splenomegaly may improve portal hypertension (pHTN), ruxolitinib was evaluated in 21 patients with MPN-SVT (12 MF, 5 PV, 4 ET). Twenty-nine percent experienced ≥35% spleen volume reduction, and 4 evaluable patients experienced a 24% reduction in spleen stiffness, but no significant improvements in esophageal varices were observed.64 In patients with MPN-SVT who lack cytosis, the impact of cytoreduction is unclear.61
Consequences of pHTN, including esophageal varices and refractory ascites, can necessitate transjugular intrahepatic portosystemic shunts (TIPS).52 In a retrospective study of 29 patients with MPN, TIPS was effective in resolving pHTN and its consequences, but 31% experienced in-stent thrombosis requiring additional interventions.65 Finally, in patients with BCS with progressive liver dysfunction, transplantation may be required.52 Among 78 patients with BCS who underwent liver transplantation, 41 had an MPN (40 with ET or PV) and had long-term survival comparable to that of those without MPN.66 Although 24% experienced bleeding or thrombosis (fatal in 4 patients, recurrent BCS in 2 patients), immunosuppression did not accelerate MPN progression.66
Bleeding risk
Bleeding in MPN is often multifactorial, with some risk factors being intuitive and others inconsistent across studies (Table 1).3,4,67,68 Regarding MPN-specific factors, thrombocytosis affects bleeding risk. In a longitudinal cohort study of ET (N = 776), a platelet count above normal range in follow-up conferred an increased risk of bleeding (HR, 3.7; 95% CI, 1.7-8.2).14 Leukocytosis also independently increased risk of bleeding in a study of early primary myelofibrosis and ET (HR, 1.74; P = .04),3 and a U-shaped relationship between leukocytes and risk of hemorrhage was found in a longitudinal cohort study of patients with ET, with hazard of bleeding rising together with leukocyte count.14 A meta-analysis including patients with ET and PV reported an association between leukocytosis and bleeding (RR, 1.87; 95% CI, 1.59-2.23).15
Risk factors for myeloproliferative neoplasm bleeding
| Risk factor | Comment |
|---|---|
| Advanced age | Prospective PV cohort,68 retrospective study of all MPN subtypes67 |
| Disease duration | Prospective PV cohort68 |
| Splenomegaly | Odds ratio 2.24 in German prospective cohort |
| Prior thrombosis | Odds ratio 2.74 in German prospective cohort |
| Portal HTN | As in BCS and PVT, or with massive splenomegaly or hepatic extramedullary hematopoiesis |
| Prior hemorrhage | WHO-defined ET and PMF,3 as well as PV68 |
| MPN subtype | MF > ET/PV |
| Driving mutation | JAK2 V617F may associate with aVWS71 in ET |
| Unclear if risk is greater in CALR vs JAK2 V617F-positive MPNs | |
| Thrombocytosis | With or without aVWS |
| Leukocytosis | ET and early MF3 |
| aVWS | Identified even in absence of extreme thrombocytosis71 |
| Treatment | Antiplatelet therapy? |
| Anticoagulation? | |
| Antiplatelet therapy with anagrelide41 |
| Risk factor | Comment |
|---|---|
| Advanced age | Prospective PV cohort,68 retrospective study of all MPN subtypes67 |
| Disease duration | Prospective PV cohort68 |
| Splenomegaly | Odds ratio 2.24 in German prospective cohort |
| Prior thrombosis | Odds ratio 2.74 in German prospective cohort |
| Portal HTN | As in BCS and PVT, or with massive splenomegaly or hepatic extramedullary hematopoiesis |
| Prior hemorrhage | WHO-defined ET and PMF,3 as well as PV68 |
| MPN subtype | MF > ET/PV |
| Driving mutation | JAK2 V617F may associate with aVWS71 in ET |
| Unclear if risk is greater in CALR vs JAK2 V617F-positive MPNs | |
| Thrombocytosis | With or without aVWS |
| Leukocytosis | ET and early MF3 |
| aVWS | Identified even in absence of extreme thrombocytosis71 |
| Treatment | Antiplatelet therapy? |
| Anticoagulation? | |
| Antiplatelet therapy with anagrelide41 |
aVWS, acquired von Willebrand syndrome; BCS, Budd-Chiari syndrome; ET, essential thrombocythemia; HTN, hypertension; MF, myelofibrosis; MPN, myeloproliferative neoplasm; PMF, primary myelofibrosis; PV, polycythemia vera; PVT, portal vein thrombosis; WHO, World Health Organization.
Bleeding risk may be worsened by the development of acquired von Willebrand syndrome (aVWS), which may result from increased proteolysis with platelet activation leading to reduced von Willebrand factor activity.69,70 Noting possible selection bias, one study evaluated 116 patients with ET and 57 patients with PV for aVWS.71 Sixty-four (55%) patients with ET and 28 (49%) patients with PV met criteria (including ristocetin activity <41% and <58% among O and non-O blood groups, respectively), with a median ristocetin activity of 35%.71 Interestingly, there was no difference in the rate of aVWS when comparing platelet counts of 500 × 109/L to 1000 × 109/L vs >1000 × 109/L (P = .43). In fact, most (69.5%) patients with aVWS had platelet counts <1000 × 109/L. Furthermore, most patients with major bleeding had platelet counts <1000 × 109/L.71 In ET, those with JAK2 V617F were more likely to develop aVWS than CALR-ET or triple-negative ET (70.3 vs 45.7%, P = .02; 70.3% vs 17.6%, P < .001).71 This study suggests consideration of aVWS even in the absence of extreme thrombocytosis.
Apart from a possible association of JAK2 V617F with aVWS, driver mutations may not affect bleeding risk in the absence of antithrombotic therapy. Though CALR associates more with thrombocytosis, in one retrospective study, CALR mutational status vs JAK2 V617F did not predict bleeding.67 Studies of JAK2 V617F allele burden have been conflicting. One found the prevalence and incidence rate (IR) of hemorrhage to correlate with higher allele burden (IR increased from 0.7/100 patient-years for first quartile to 3.23/100 for fourth quartile [IR ratio, 4.6]),72 whereas another study did not find any association between allele burden >20% and bleeding risk.73
MPN therapy can also impact bleeding rates. The use of low-dose antiplatelet therapy did not affect bleeding risk in the European Collaboration on Low-Dose Aspirin in Polycythemia Vera study of patients with PV.31 In the meta-analysis of ET studies, whereas RRs for antiplatelet-associated bleeding were possibly increased, the evidence was considered inconsistent and at high risk of bias.32 Combining aspirin with ANA may increase risk, owing to synergistic interference of platelet function; serious bleeding rates were higher for those who received ANA/aspirin than in those who received HU/aspirin (OR, 2.61; 95% CI, 1.27-5.33) in a randomized study, and, similarly, in a large observational cohort study, higher rates were found with ANA/antiplatelet therapy than with cytoreductive/antiplatelet therapy (IR, 1.35 vs 0.33 events per 10 000 patient-years of exposure, respectively).74,75 The effect of antithrombotic therapy on bleeding has been inconsistent. VKA and acetylsalicylic acid did not increase risk of bleeding in a German registry study.4 Similarly, a study of 150 patients with ET/PV treated with VKA found no difference in major bleeding rates for periods on VKA compared with off-VKA (IR, 1.8 vs 1.5 per 100 patient-years).76 In contrast, a study of 206 patients with MPN-related VTE found a trend toward a higher rate of major bleeding on VKA (IR, 2.4/100 patient-years) vs off VKA (IR, 0.7/100 patient-years) (P = .08).77
Treatment considerations
Management can include antifibrinolytic agents and platelet transfusions, desmopressin and factor replacement (and cytoreduction) if aVWS is present, and cytoreduction in those with recurrent unexplained bleeding episodes, even in the absence of elevated blood counts or high thrombotic risk profile.78 Management of extreme thrombocytosis is heterogeneous. When hematologists, including 57% considered MPN specialists, were surveyed about extreme thrombocytosis scenarios, there was no consensus about threshold platelet counts triggering cytoreduction in low-risk ET, extreme thrombocytosis as a treatment criterion in PV, platelet goal in cytoreduction for extreme thrombocytosis, and whether aVWS testing should guide aspirin use.79
Surgery
Patients with MPN undergoing surgery are at high risk for both thrombosis and bleeding.9 A retrospective study of 311 (155 major, 156 minor) surgical procedures in patients with PV/ET found high rates of vascular occlusion (7.7%; estimated 5-fold deep vein thrombosis risk) and major hemorrhage (7.3%) despite antithrombotic prophylaxis, blood count control (mean Hct, 42.7%; leukocyte count, 9.5 × 109/L; platelet count, 506 × 109/L), and use of cytoreduction and phlebotomy (74%).80 Arterial thrombosis was more frequent in ET (HR, 3.3) and in those with arterial risk factors, whereas venous events were more common in PV (HR, 7.3).80 NCCN provides guidance on perioperative management, summarized in Figure 4.49
Pregnancy
Most information on pregnancy in MPN is based on patients with ET. In a prospective study including 58 pregnancies (81% ET, 9% PV), 88% of women received aspirin, 38% received LMWH, and 14% were treated with IFN.81 Preeclampsia was the most common antepartum complication (9%). The miscarriage rate was 1.7/100, and the perinatal mortality was 17/1000; 23% of neonates had low birth weights. The cesarean section rate was 45%, and although 9% (3.5% major) experienced postpartum hemorrhage, no thrombotic events were reported.81 This study could not identify specific risk factors for complications. Results of a retrospective cohort study of 27 pregnancies (19 pregnancies in 9 with ET, 8 pregnancies in 5 with PV) are less encouraging; that study reported a cesarean section rate of 70% and only a 30% rate of uneventful pregnancies.82 Complications included early miscarriage (22%), growth restriction (15%), premature delivery (15%), placental dysfunction (15%), thrombosis (15%), and hemorrhage (11%).82 Among the 18 pregnancies that were considered high risk, most commonly due to prior thrombosis, treatments included aspirin (n = 10), LMWH (n = 8), and LMWH with IFN (n = 5). In a separate study of 25 PV pregnancies, the live birth rate was 62.5%, and the maternal complication rate was 16.7%. Late fetal losses (16.7%) were noted, but less frequently in those managed with antithrombotic therapy (n = 19) vs untreated patients (n = 5) (10.5% vs 40%). All women were phlebotomized, but only 1 patient was managed according to CYTO-PV thresholds.83
Management remains empirical, and no treatment is proven to affect outcomes, though aspirin, LMWH, and IFNs are considered, as is phlebotomy in PV, to maintain Hct within the normal range for gestation (first trimester, <41%; second trimester, <38%; third trimester, <39%) (Figure 4).84 A meta-analysis of 756 ET pregnancies identified an antepartum VTE risk of 2.5%, which was below a threshold (>3%) to show clear benefit of LMWH prophylaxis in otherwise low-risk patients.85 However, the postpartum VTE risk of 4.4% supports prophylaxis after delivery. Aspirin improved live birth rates (OR, 5.0) in ET in a meta-analysis of 1226 MPN pregnancies, though LMWH did not.86 IFN also increased the live birth rate (OR, 3.9)86 and is recommended by some in women with prior pregnancy loss or complications.84 It is noteworthy, however, that the majority of studies were retrospective, spanned 4 decades, and included small sample sizes.86 Prospective, controlled studies are needed to solidify treatment recommendations.
Case follow-up
The patient underwent paracentesis and TIPS and started anticoagulation with warfarin; aspirin was added after subsequent TIPS occlusion. Leukocytosis and thrombocytosis improved, possibly masked by hepatic insult and hypersplenism. Her MPN initially remained unclassifiable, but after 4 years, erythrocytosis emerged (hemoglobin, 16.5 g/dL), and thrombocytosis worsened (695 × 109/L), prompting phlebotomy and HU. Owing to erratic international normalized ratios, she was transitioned to a DOAC. She undergoes surveillance endoscopy and magnetic resonance imaging, which last revealed mild portal gastropathy and hepatomegaly with regenerative nodules. She has contemplated another pregnancy, which would require LMWH and pegylated IFN.
Conclusions
Management of MPN thrombosis and bleeding remains challenging. Understanding the multifactorial contributions will lead to more precise risk classification and treatment strategies. Recent work highlights the contribution of inflammation, as well as the interaction between JAK2 V617F and endothelial cells, leading to activation, abnormal leukocyte adhesion, and increase in thrombosis risk. Furthermore, JAK2 V617F may lead to NET formation and unstable atherosclerotic plaques, contributing to vascular occlusion. Future goals include incorporation of relevant biomarkers of inflammatory stress and abnormal adhesion; CRP, PTX3, and P-selectin are potential candidates pending prospective study. In addition, future evaluations of novel agents in ET/PV should include thrombosis reduction as a primary endpoint because complete hematological response is not a surrogate for thrombosis reduction. Although surrogates for thrombosis may be helpful, prior landmark studies12,31,74 have proved that thrombosis reduction is a feasible endpoint, and a recent meta-analysis provides risk estimates that will be useful for sample size calculation.5 Future treatment strategies targeting inflammation and abnormal adhesion may reduce recurrent thrombosis, which remains a problem, especially for those with SVT and MF. Management of SVT and other special situations remains largely empirical and consensus based. However, with the rapid pace of discovery, it is hoped that precise risk assessment and tailored therapies will help hematologists reduce the burden of thrombohemorrhagic complications in MPN.
Authorship
Contribution: B.L.S. and K.M. reviewed the medical literature, synthesized the review, and drafted the manuscript together.
Conflict-of-interest disclosure: B.L.S. has served on advisory boards for Incyte Corporation, Celgene, and Apexx Oncology. K.M. declares no competing financial interests. Off-label drug use: None disclosed.
Correspondence: Brady L. Stein, Division of Hematology/Oncology and Robert H. Lurie Comprehensive Cancer Center, Department of Medicine, Northwestern University Feinberg School of Medicine, 645 N Michigan Ave, Suite 1020, Chicago, IL 60611; e-mail: bstein@nm.org.
This article was selected by the Blood and Hematology 2019 American Society of Hematology Education Program editors for concurrent submission to Blood and Hematology 2019. It is reprinted in Hematology Am Soc Hematol Educ Program. 2019;2019:397-406.
