

Key Points
MELK promotes EZH2 stability through phosphorylation-dependent loss of ubiquitination.
Overexpressed MELK mediates sensitivity to bortezomib through modulating EZH2 stability in NKTL.

Abstract
Oncogenic EZH2 is overexpressed and extensively involved in the pathophysiology of different cancers including extranodal natural killer/T-cell lymphoma (NKTL). However, the mechanisms regarding EZH2 upregulation is poorly understood, and it still remains untargetable in NKTL. In this study, we examine EZH2 protein turnover in NKTL and identify MELK kinase as a regulator of EZH2 ubiquitination and turnover. Using quantitative mass spectrometry analysis, we observed a MELK-mediated increase of EZH2 S220 phosphorylation along with a concomitant loss of EZH2 K222 ubiquitination, suggesting a phosphorylation-dependent regulation of EZH2 ubiquitination. MELK inhibition through both chemical and genetic means led to ubiquitination and destabilization of EZH2 protein. Importantly, we determine that MELK is upregulated in NKTL, and its expression correlates with EZH2 protein expression as determined by tissue microarray derived from NKTL patients. FOXM1, which connected MELK to EZH2 signaling in glioma, was not involved in mediating EZH2 ubiquitination. Furthermore, we identify USP36 as the deubiquitinating enzyme that deubiquitinates EZH2 at K222. These findings uncover an important role of MELK and USP36 in mediating EZH2 stability in NKTL. Moreover, MELK overexpression led to decreased sensitivity to bortezomib treatment in NKTL based on deprivation of EZH2 ubiquitination. Therefore, modulation of EZH2 ubiquitination status by targeting MELK may be a new therapeutic strategy for NKTL patients with poor bortezomib response.
Introduction
EZH2 is the enzymatic subunit of the polycomb repressive complex 2 (PRC2), which specifically methylates histone H3 at lysine 27 and thus mediates gene expression repression. As a histone methyltransferase, EZH2 needs to assemble with other PRC2 components such as SUZ12 and EED to fulfill its catalytic activity. EZH2 was found to be overexpressed in different cancer types, including extranodal natural killer/T-cell lymphoma (NKTL),1 with a well-established oncogenic role in these cancers. Overexpressed EZH2 associates with tumor initiation and progression as well as poor clinical outcomes. And EZH2 overexpression also attenuates sensitivity to chemotherapy regimens in different cancers.2-4
However, the mechanisms underlying EZH2 overexpression in cancers are complicated, context-dependent, and multifaceted. Studies in recent years suggest that transcriptional activation, microRNA (miR) deregulation and deregulated proteolytic system are the 3 main causes leading to its overexpression. EZH2 is transactivated by HIF-1α under hypoxia in breast cancer-initiating cells5 and by E2F1 in bladder cancer.6 Frequent loss of miR-101, which transcriptionally repress EZH2 expression, was first identified in localized and metastatic prostate cancer7 and reported in other tumors.8 EZH2 oncoprotein could be stabilized by aberrant upregulation of SKP2, which inhibited TRAF6-mediated EZH2 ubiquitination,9 or through the deubiquitination of upregulated USP2110 in urological cancers. In NKTL, our previous study showed that the expression of miR-101 and miR-26a/b inversely correlated with EZH2, and an attenuation of EZH2 expression was observed with lentiviral transduction of these miRs.1
Most commonly, EZH2 exerts its tumorigenic function through H3K27 trimethylation-dependent gene-silencing machineries. Whereas in a few malignant cancer contexts, such as NKTL1 and triple-negative breast cancer,11 EZH2 may exhibit tumorigenicity as a transcriptional activator that is PRC2- and H3K27 trimethylation-independent. Since 2013, a series of small-molecule inhibitors as well as peptide inhibitor of EZH2 have been developed. All these inhibitors deplete H3K27 trimethylation level, leaving EZH2 protein intact, and thus cannot effectively target oncogenic EZH2 that is independent of its histone modifying function in NKTL. Therefore, it is important to look at other mechanisms that affect EZH2 protein levels, including the turnover of EZH2 in NKTL.
MELK is a highly conserved AMPK-family serine/threonine kinase that mediates signals initiated by exogenous stimulus. MELK is overexpressed in a wide panel of cancers that catalyzes phosphorylation on oncogenic FOXM1.12 In this study, we demonstrate that in NKTL, overexpressed MELK promotes the stability of EZH2 protein through phosphorylation- and ubiquitination-coupled machineries. MELK specifically phosphorylates EZH2 at S220, resulting in decreased levels of K222 EZH2 ubiquitination. These findings uncover a novel mechanism explaining EZH2 overexpression in NKTL and identify MELK kinase upregulation as a novel biomarker for response in NKTL patients receiving the first therapeutic proteasome inhibitor bortezomib.
Materials and methods
Cell culture
The NKTL cell lines used included NKYS, KHYG-1, NK-S1, HANK-1, SNK1, SNK6, and NK-92. NK-92 cells were cultured in α-MEM without nucleotides (Thermo Fisher Scientific) supplemented with 12.5% fetal bovine serum (Biowest), 12.5% horse serum (Biowest), 0.1 mM 2-mercaptoethanol, and 100U/mL of interleukin-2 (Miltenyi). Culture conditions for all the other NKTL cell lines used in this study have been described previously.13,14 Detailed characteristics of these NKTL cell lines were described (supplemental Table 1 on the Blood Web site). Normal NK cells were isolated from human peripheral blood of healthy donors (from Dario Campana, National University of Singapore).15 Isolated normal NK cells were cultured in CellGenix GMP SCGM medium supplemented with 200 U/mL of interleukin-2 (Miltenyi). HEK293 cells and HEK293T cells were both cultured in Dulbecco's modified Eagle medium (Biowest) supplemented with 10% fetal bovine serum (Biowest).
Drugs and antibodies
See supplemental Methods for details.
Constructs, shRNA and siRNA transfection
For NKTL cell lines, transfections of overexpressing constructs, short hairpin (shRNA) or small interfering RNA (siRNA) were performed using the Neon Transfection system (Life Technology) following the manufacturer’s protocols. For HEK293 cells or HEK293T cells, the X-tremeGene HP DNA Transfection Reagent (Roche 6366546001) was used for constructs or shRNA transfections following the manufacturer’s protocol. Details of constructs, shRNA and siRNA used in this study are available in supplemental Methods.
Cell survival assay
The cell survival was measured using the CellTiter-Glo luminescent cell viability assay kit (Promega G7573) according the manufacturer’s indications.
Nuclear-cytoplasmic fractionation
The NE-PER Nuclear and Cytoplasmic Extraction Reagents (Thermo Fisher Scientific) were used for the fractionation based on the manufacturer’s protocols. Both the cytoplasmic and nuclear lysates, cytoplasm alone or nucleus alone were used for coimmunoprecipitation (co-IP) or immunoblots.
Nucleoplasm and nucleolus extraction
Sequential cell lysis was used to obtain nucleoplasm and nucleolus extracts. See supplemental Methods for details.
Co-IP and western blotting
For co-IP using HEK293T cells or NKTL cells, the cell lysates were prepared using RIPA buffer plus protease inhibitor (Roche), and were subject to sonication (Active motif Probe Sonicator; 30% amplitude, 30-second on and 30-second off bursts, 2 cycles) before pull-down. After clarification by centrifugation and protein concentration determination, antibodies were added in to the lysate and the lysates were rotated at 4°C overnight. The protein A/G beads (Thermo Fisher Scientific 10004G) were then added (25 µL/10 million cells), followed by incubation at 4°C for 1 hour. After washing with lysis buffer, 2× Laemmli buffer (Bio-Rad) with 5% β-mercaptoethanol were added to the beads, which then were boiled at 95°C for 5 minutes. For western blotting, the cell lysates were prepared using RIPA buffer plus protease inhibitor (Roche), but were subject to sonication for 30-second on, 30-second off bursts (2 cycles) using Bioruptor (Diagenode). Then the lysates were spun down and the protein concentration was calculated. The supernatants were similarly mixed with Laemmli buffer and boiled. The boiled sample were resolved in sodium dodecyl sulfate-polyacrylamide gel electrophoresis and immunoblotted with indicated antibodies.
Quantitative reverse-transcription PCR
Total RNA was extracted using RNeasy Mini Kit (Qiagen) and reverse-transcribed using the iScript Reverse Transcription Supermix for quantitative reverse transcription polymerase chain reaction (PCR; Bio-Rad) (25°C 5 minutes; 42°C 30 minutes; 85°C 5 minutes). The reaction was performed on a QuantStudio Real-Time PCR System (Thermo Fisher Scientific) using SYBR Green Mix (Bio-Rad). Primers for the reactions are available in supplemental Methods.
Immunofluorescence staining and analysis
NKTL patient biopsies were stained in tissue microarray format. Refer to supplemental Methods for details of staining and analysis procedures.
SILAC-based affinity purification for MS
For stable isotope labeling with amino acids in cell culture (SILAC) labeling, HEK293T cells were incubated in DMEM (-Arg, -Lys) medium containing 10% dialyzed fetal bovine serum (Thermo Fisher Scientific) supplemented with 42 mg/L 13C615N4l-arginine and 73 mg/L 13C615N2l-lysine (Cambridge Isotope; “heavy”) or the corresponding non-labeled amino acids (“light”), respectively. Successful SILAC incorporation was verified by in-gel trypsin digestion and mass spectrometry (MS) analysis of heavy input samples in parallel to IP samples to ensure adequate incorporation. The “light” population were transfected with all lysine-mutated ubiquitin (K0-ub) plus EZH2 but without MELK, and the “heavy” population were transfected with K0-ub plus EZH2 and MELK (“forward” pull-down). A label-swap (“reverse”) replicating experiment was also performed. After 48 hours, cells were harvested and lysed in RIPA buffer with protease inhibitor, followed by co-IP with EZH2 antibody for pull-down. The protein lysates were separated by sodium dodecyl sulfate-polyacrylamide gel electrophoresis, and then the gel was stained, excised, and processed for MS analysis. Details of MS procedures and data analysis are available in supplemental Methods and the MS data has been deposited to the ProteomeXchange Consortium via PRIDE16 (PXD015008).
Kinase assay coupled with MS
The 15-aa peptide E1 (RPPRKFPSDKIFEAI) and EZH2 S220 phosphorylation signal (p-E1)[RPPRKFPS(p)DKIFEAI] corresponding to S220 phosphorylation site on EZH2 were designed and synthesized (Bio basic). Both E1 and p-E1 were preloaded to liquid chromatography MS/MS system as standards before kinase assay. Details of the kinase reaction and MS parameters are available in the supplemental Methods.
Results
Overexpressed MELK interacts with and stabilizes EZH2
As identified by gene expression profiling, MELK is one of the significantly overexpressed genes in NKTL patient samples and cell lines (Figure 1A). We then validated MELK overexpression in a panel of NKTL cell lines. Compared with normal NK cells, all NKTL cell lines tested displayed increased levels of MELK expression. And general expression correlation between MELK and EZH2 was observed in NKTL cell lines (Figure 1B).

Overexpressed MELK correlates with EZH2 expression in NKTL. (A) Gene expression profiling data showing MELK expression in NKTL cell lines and patient samples of NKTL (extranodal) and EBV+ peripheral T-cell lymphoma (nodal) cases. (B) Expression of MELK and EZH2 in normal NK and a panel of NKTL cell lines. Densitometry analysis was used to quantify average changes in 3 individual experiments. (C) IP showing MELK-EZH2 interaction. (D-E) EZH2 protein level change with MELK inhibitor OTSSP167 treatment of 48 hours in NKYS (D, densitometry analysis was used to quantify average changes in 3 individual experiments) and (E) NK-S1. (F-G) EZH2 protein level change with MELK knock-down using siRNA in (F) NKYS and (G) NK-S1. Cells were harvested for immunoblots 48 hours after knock-down. (H) Linear correlation was obtained by comparing percentage of EZH2 positive staining (EZH2+) to average percentage of cytoplasmic and nuclear MELK positive staining (MELK+) in NKTL cells (CD3+) for each core from NKTL patient tissues. N = 83 cores from 52 NKTL patients were stained (R = 0.4549, P < .0001). (I) Representative images indicating expression of EZH2 and MELK in NKTL tissue microarray sample (left) with corresponding segmented image masks (right). CD3 marks tumor cells. A total of 80.8% of CD3+ cells in sample 1 are positive for nuclear expression of EZH2, whereas 38.27% CD3+ cells in sample 2 are positive for EZH2. Seventy-three percent of CD3+ cells in sample 1 are positive for MELK and only 36.42% CD3+ cells are positive for MELK in sample 2. All immunoblots were performed in at least 3 individual experiments; representative images are shown. Ig, immunoglobulin
Overexpressed MELK correlates with EZH2 expression in NKTL. (A) Gene expression profiling data showing MELK expression in NKTL cell lines and patient samples of NKTL (extranodal) and EBV+ peripheral T-cell lymphoma (nodal) cases. (B) Expression of MELK and EZH2 in normal NK and a panel of NKTL cell lines. Densitometry analysis was used to quantify average changes in 3 individual experiments. (C) IP showing MELK-EZH2 interaction. (D-E) EZH2 protein level change with MELK inhibitor OTSSP167 treatment of 48 hours in NKYS (D, densitometry analysis was used to quantify average changes in 3 individual experiments) and (E) NK-S1. (F-G) EZH2 protein level change with MELK knock-down using siRNA in (F) NKYS and (G) NK-S1. Cells were harvested for immunoblots 48 hours after knock-down. (H) Linear correlation was obtained by comparing percentage of EZH2 positive staining (EZH2+) to average percentage of cytoplasmic and nuclear MELK positive staining (MELK+) in NKTL cells (CD3+) for each core from NKTL patient tissues. N = 83 cores from 52 NKTL patients were stained (R = 0.4549, P < .0001). (I) Representative images indicating expression of EZH2 and MELK in NKTL tissue microarray sample (left) with corresponding segmented image masks (right). CD3 marks tumor cells. A total of 80.8% of CD3+ cells in sample 1 are positive for nuclear expression of EZH2, whereas 38.27% CD3+ cells in sample 2 are positive for EZH2. Seventy-three percent of CD3+ cells in sample 1 are positive for MELK and only 36.42% CD3+ cells are positive for MELK in sample 2. All immunoblots were performed in at least 3 individual experiments; representative images are shown. Ig, immunoglobulin
To examine the potential association between MELK and EZH2, we pulled down endogenous EZH2 in NKTL cells and found EZH2 interacted with MELK under physiological conditions (Figure 1C). Because several recent studies have uncovered a role of MELK in regulating EZH2 level,17,18 we treated NKTL cells with the MELK inhibitor OTSSP167 (OTS) to check whether MELK also modulated EZH2 expression in NKTL. With OTS treatment, a clear reduction in EZH2 protein levels could be seen along with a reduction of FOXM1 and p-FOXM1 (Figure 1D-E). Whereas overall EZH2 messenger RNA (mRNA) levels did not decrease correspondingly (supplemental Figure 1A-B). Similarly, knock-down of MELK resulted in a significant downregulation of EZH2 protein but not EZH2 mRNA (Figure 1F-G; supplemental Figure 1C-D). Interestingly, cytoplasmic and nuclear MELK displayed a similar level of expression (supplemental Figure 2A) with MELK interacting with EZH2 in both cytoplasm and nucleus (supplemental Figure 2B). Next, we analyzed the expression of MELK and EZH2 in tissue samples derived from NKTL patients and observed MELK-EZH2 correlation (Figure 1H-I; N = 83 cores from 52 NKTL patients; R=0.4549; P<.0001). These data suggest that MELK associates with EZH2 and promotes EZH2 protein stability in NKTL.
MELK upregulates EZH2 level by inhibiting its ubiquitination
Given that MELK could stabilize EZH2 protein, we investigated whether MELK participated in the regulation of EZH2 ubiquitination. We overexpressed MELK and observed a clear reduction in EZH2 ubiquitination (Figure 2A). Conversely, MELK knock-down or MELK inhibitor treatment resulted in a significant increase in EZH2 ubiquitination (Figure 2B-C). These observations suggest that MELK is involved in inhibiting EZH2 protein ubiquitination.

MELK mediates ubiquitination of EZH2. (A) Change of EZH2 ubiquitination level with MELK overexpression in HEK293T cells. Before cell harvesting, MG132 was given at 10 µM for 6 hours of treatment 48 hours after transfection. (B) Change of EZH2 ubiquitination level with MELK knock-down in NKYS cells. Forty-eight hours after transfection, 1 µM of MG132 was used for treatment of 6 hours before cell harvesting. (C) Change of EZH2 ubiquitination level with OTSSP167 treatment in NKYS cells. A total of 10 nM of OTSSP167 and 2 µM of MG132 was used for 6 hours’ treatment before harvesting. Densitometry analysis was used to quantify average changes in 3 individual experiments. (D) Schematic protocol of SILAC-MS experiment. (E) Expression level of HA-tagged EZH2 wild-type and ubiquitination-dead mutants in NKTL cell lines. The transfection was performed using electroporation. Cells were harvested for immunoblots 24 hours after transfection. (F) IP showing the change of ubiquitination level for EZH2 wild-type or mutants in HEK293T cells. Before cell harvesting, MG132 was given at 5 µM for 6 hours of treatment 48 hours after transfection. Densitometry analysis was used to quantify average changes in 3 individual experiments. (G) Change of interaction between MELK and EZH2 wild-type or its mutants in HEK293T cells. Cells were harvested for co-IP 48 hours after transfection. All immunoblots were performed in at least 3 individual experiments and representative images are shown. CON, control; EV, empty vector; GAPDH, glyceraldehyde-3-phosphate dehydrogenase; HA, hemagglutinin; LC, liquid chromatography; Ub, ubiquitination; WT, wild-type.
MELK mediates ubiquitination of EZH2. (A) Change of EZH2 ubiquitination level with MELK overexpression in HEK293T cells. Before cell harvesting, MG132 was given at 10 µM for 6 hours of treatment 48 hours after transfection. (B) Change of EZH2 ubiquitination level with MELK knock-down in NKYS cells. Forty-eight hours after transfection, 1 µM of MG132 was used for treatment of 6 hours before cell harvesting. (C) Change of EZH2 ubiquitination level with OTSSP167 treatment in NKYS cells. A total of 10 nM of OTSSP167 and 2 µM of MG132 was used for 6 hours’ treatment before harvesting. Densitometry analysis was used to quantify average changes in 3 individual experiments. (D) Schematic protocol of SILAC-MS experiment. (E) Expression level of HA-tagged EZH2 wild-type and ubiquitination-dead mutants in NKTL cell lines. The transfection was performed using electroporation. Cells were harvested for immunoblots 24 hours after transfection. (F) IP showing the change of ubiquitination level for EZH2 wild-type or mutants in HEK293T cells. Before cell harvesting, MG132 was given at 5 µM for 6 hours of treatment 48 hours after transfection. Densitometry analysis was used to quantify average changes in 3 individual experiments. (G) Change of interaction between MELK and EZH2 wild-type or its mutants in HEK293T cells. Cells were harvested for co-IP 48 hours after transfection. All immunoblots were performed in at least 3 individual experiments and representative images are shown. CON, control; EV, empty vector; GAPDH, glyceraldehyde-3-phosphate dehydrogenase; HA, hemagglutinin; LC, liquid chromatography; Ub, ubiquitination; WT, wild-type.
Next, to confirm the role of MELK in EZH2 ubiquitination and to identify the corresponding ubiquitination site(s) altered by MELK expression, we immunoprecipitated EZH2 from SILAC-labeled HEK293T cells cotransfected with all-lysine-mutated ubiquitin (K0-ub), EZH2, and either MELK or an empty vector control. We performed these experiments in a “forward” reaction with MELK overexpressed in the “heavy” labeled cells and the empty vector control used in “light” labeled cells, as well as a reciprocal “reverse” reaction with exchanged SILAC labels (Figure 2D). The MS analysis identified 2 possible sites of EZH2 ubiquitination: K222 and K629 (supplemental Table 2).
K222 is the critical site of EZH2 ubiquitination
We generated ubiquitination-null mutants (EZH2 K222R, EZH2 K629R) to verify the site of EZH2 ubiquitination suggested by the MS data. In NKTL cells, a stabilization of EZH2 level could be observed with K222R but not K629R mutant compared with EZH2 WT (Figure 2E). Moreover, both K222 and K629 mutants showed attenuated interaction with MELK (Figure 2G), indicating that these 2 sites are important for MELK-EZH2 association, whereas only the K222R mutant displayed decreased level of EZH2 ubiquitination (Figure 2F). Therefore, K222 is the critical site of EZH2 ubiquitination affected by MELK.
MELK mediates EZH2 ubiquitination linked to site-specific phosphorylation
Considering that MELK itself is a serine/threonine kinase, it is probable that MELK mediates EZH2 ubiquitination following phosphorylation on EZH2. Thus, we reanalyzed the SILAC-MS data for all the potential sites of EZH2 phosphorylation, and S220 phosphorylation was the only one that depended on MELK activity.
Interestingly, the S220 phosphorylation site is very close to the K222 ubiquitination site based on EZH2 3-dimensional structure,19 suggesting the phosphorylation may be a determinant for the inhibition of ligase-mediated ubiquitination (Figure 3A). MELK-mediated EZH2 S220 phosphorylation was further verified through in vitro kinase assay (supplemental Figure 3). As indicated, a clear peak corresponding to EZH2 S220 phosphorylation signal (p-E1) was visualized when recombinant active MELK was incubated with reaction mixture at 30°C for 60 minutes. Almost no S220 phosphorylation was observed without incubation (Figure 3B-C). These data show that MELK directly phosphorylates EZH2 at S220.
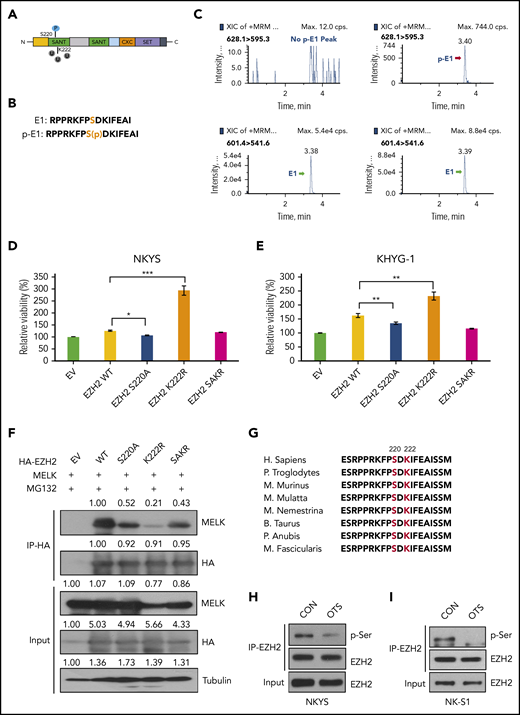
MELK affects EZH2 ubiquitination following site-specific phosphorylation. (A) Schematic model showing localizations of S220 phosphorylation site and K222 ubiquitination site on EZH2 protein. (B) Amino acid sequence of E1 and p-E1 peptides. (C) In vitro kinase assay showing MELK could phosphorylate EZH2 at S220. (D-E) Different ratio of cell survival for (D) NKYS and (E) KHYG-1 cells transfected with empty vector, EZH2 wild-type or mutant plasmids. NKYS and KHYG-1 cells were transfected with indicated plasmids through electroporation and subject to overnight puromycin selection (1 µg/mL) 7 hours after transfection. Cell survival was measured using CellTiter-Glo reagent. (F) Change of interaction between MELK and EZH2 wild-type or mutants in HEK293T cells. Cells were harvested for co-IP 48 hours after transfection. Densitometry analysis was used to quantify average changes in 3 individual experiments. (G) Schematic diagram indicating the conservation of EZH2 S220 and K222. (H-I) Change of EZH2 total serine phosphorylation upon 16 hours of OTSSP167 treatment in NKYS (H, 10 nM) and NK-S1 (I, 20 nM) cells. All immunoblots were performed in at least 3 individual experiments; representative images are shown.
MELK affects EZH2 ubiquitination following site-specific phosphorylation. (A) Schematic model showing localizations of S220 phosphorylation site and K222 ubiquitination site on EZH2 protein. (B) Amino acid sequence of E1 and p-E1 peptides. (C) In vitro kinase assay showing MELK could phosphorylate EZH2 at S220. (D-E) Different ratio of cell survival for (D) NKYS and (E) KHYG-1 cells transfected with empty vector, EZH2 wild-type or mutant plasmids. NKYS and KHYG-1 cells were transfected with indicated plasmids through electroporation and subject to overnight puromycin selection (1 µg/mL) 7 hours after transfection. Cell survival was measured using CellTiter-Glo reagent. (F) Change of interaction between MELK and EZH2 wild-type or mutants in HEK293T cells. Cells were harvested for co-IP 48 hours after transfection. Densitometry analysis was used to quantify average changes in 3 individual experiments. (G) Schematic diagram indicating the conservation of EZH2 S220 and K222. (H-I) Change of EZH2 total serine phosphorylation upon 16 hours of OTSSP167 treatment in NKYS (H, 10 nM) and NK-S1 (I, 20 nM) cells. All immunoblots were performed in at least 3 individual experiments; representative images are shown.
Next, we generated EZH2 phosphorylation-dead (S220A) and double (S220A K222R: SAKR) mutants. Because EZH2 is oncogenic in NKTL,1 any alteration of EZH2 stability will naturally affect NKTL cell survival. As anticipated, the EZH2 K222R mutant could further promote cell survival compared with EZH2 wild-type, and both the S220A and SAKR mutant compromised the survival advantage conferred by EZH2 wild-type transfection (Figure 3D-E). All of these EZH2 mutants displayed attenuated interaction with MELK (Figure 3F). The similar effects displayed by S220A and SAKR mutants also reinforce that S220 phosphorylation may prime EZH2 for K222 ubiquitination. Notably, both EZH2 S220 and K222 are evolutionarily conserved sites (Figure 3G).
Moreover, MELK-mediated EZH2 serine phosphorylation was also validated in NKTL cells. When we treated NKTL cells with MELK inhibitor OTS, we observed a decrease of EZH2 serine phosphorylation (Figure 3H-I).
FOXM1 may not be engaged in modulating EZH2 ubiquitination
Because FOXM1 serves as a bridging mediator that connects MELK to EZH2 signaling in glioma,12,17 it is necessary to investigate whether FOXM1 also plays a role in mediating EZH2 ubiquitination in NKTL. As shown in Figure 4A, FOXM1 is also overexpressed in different NKTL cell lines; however, no interaction was seen between FOXM1 and EZH2 (Figure 4B). Then we sought to examine how could FOXM1 affect EZH2 in NKTL. With FOXM1 knock-down, a decrease of EZH2 mRNA and protein level was seen in NKYS cells, which has high FOXM1 expression among all NKTL cell lines (Figure 4C,E), whereas no substantial change of EZH2 was observed in NK-S1 cells which has relatively low FOXM1 expression (Figure 4D-E). These data imply that FOXM1 is not an indispensable modulator of EZH2 in NKTL malignancy, and FOXM1 only regulates EZH2 at transcription level when it is highly overexpressed in some NKTL cell lines. Moreover, no significant alteration of EZH2 ubiquitination was seen when FOXM1 was knocked down (Figure 4F) or overexpressed (Figure 4G), and FOXM1 overexpression could not affect decrease of EZH2 ubiquitination mediated by MELK (Figure 4H).

FOXM1 may not be an upstream mediator of EZH2 ubiquitination. (A) Expression level of FOXM1 in normal NK and NKTL cell lines. (B) IP showing absence of interaction between EZH2 and FOXM1 in NKYS and NK-S1. (C-D) Quantitative reverse transcription PCR assay in (C) NKYS or (D) NK-S1 cells upon FOXM1 knock-down. Data are shown as mean ± SD, N = 3. *P < .05; **P < .01; ***P < .001. NKTL cells were harvested for RNA extraction 48 hours after transfection. (E) Immunoblots of indicated proteins upon FOXM1 knock-down in NKYS and NK-S1 cells. NKTL cells were harvested for immunoblots 48 hours after transfection. (F) Level of EZH2 ubiquitination upon knock-down of FOXM1 in NKYS cells. MG132 was given at 1 µM for 6 hours of treatment 48 hours after transfection. (G-H) Level of EZH2 ubiquitination upon overexpression of indicated proteins in HEK293T cells. MG132 was given at 10 µM for 6 hours of treatment 48 hours after transfection. (F-H) Densitometry analysis was used to quantify average changes in 3 individual experiments. All immunoblots were performed in at least 3 individual experiments and representative images are shown. SD, standard deviation.
FOXM1 may not be an upstream mediator of EZH2 ubiquitination. (A) Expression level of FOXM1 in normal NK and NKTL cell lines. (B) IP showing absence of interaction between EZH2 and FOXM1 in NKYS and NK-S1. (C-D) Quantitative reverse transcription PCR assay in (C) NKYS or (D) NK-S1 cells upon FOXM1 knock-down. Data are shown as mean ± SD, N = 3. *P < .05; **P < .01; ***P < .001. NKTL cells were harvested for RNA extraction 48 hours after transfection. (E) Immunoblots of indicated proteins upon FOXM1 knock-down in NKYS and NK-S1 cells. NKTL cells were harvested for immunoblots 48 hours after transfection. (F) Level of EZH2 ubiquitination upon knock-down of FOXM1 in NKYS cells. MG132 was given at 1 µM for 6 hours of treatment 48 hours after transfection. (G-H) Level of EZH2 ubiquitination upon overexpression of indicated proteins in HEK293T cells. MG132 was given at 10 µM for 6 hours of treatment 48 hours after transfection. (F-H) Densitometry analysis was used to quantify average changes in 3 individual experiments. All immunoblots were performed in at least 3 individual experiments and representative images are shown. SD, standard deviation.
USP36 specifically deubiquitinates EZH2
Given the spatial proximity of S220 and K222, the resulting phosphate and ubiquitin moieties may have to compete for space within the binding pocket; therefore, we speculated that K222 must be deubiquitinated to continuously balance S220 phosphorylation. As such, we sought to identify the deubiquitinase responsible for removing EZH2 ubiquitination at K222 in NKTL. USP36 and USP48 were 2 deubiquitinases that appear in our previous MS data (unpublished) in which we meant to determine proteins bound to EZH2 in NKYS cells. Using shRNA pools targeting either USP36 and USP48 (supplemental Figure 4A), we observed a decrease of EZH2 protein level (supplemental Figure 4B) as well as a corresponding increase of EZH2 total ubiquitination (supplemental Figure 4C) with only USP36 knock-down but not USP48 knock-down in HEK293T cells. Furthermore, diminished expression of USP36 significantly enhanced K48-linked polyubiquitination but not K63 polyubiquitination indicative of USP36 playing a role in EZH2 stability20,21 (supplemental Figure 4D).
As expected, ectopic expression of USP36 enhanced endogenous (supplemental Figure 4E) and exogenously expressed EZH2 levels (Figure 5A) as well as decreased EZH2 ubiquitination, which can be partially rescued upon USP36 C131A (enzymatically dead) mutant transfection (Figure 5B). In contrast, knock-down of USP36 in NKTL cells decreased EZH2 expression (Figure 5C). In line with these results, downregulation of USP36 led to a significant increase in EZH2 repressive genes expression and decrease of EZH2 activated genes expression, which we have characterized previously14 (supplemental Figure 4F). Next, we sought to establish if USP36 could interact with EZH2. As shown, endogenous EZH2 co-immunoprecipitated with GFP-USP36 (supplemental Figure 4G). Both USP36 wild-type and C131A mutant could bind to EZH2 (Figure 5D-E). Because deubiquitination effect requires both protein-protein interaction and catalyzing, such results reveal that the stabilization of EZH2 requires the enzymatic activity of USP36 besides its interaction with EZH2, reinforcing that USP36 mediates EZH2 stability through catalyzing its deubiquitination. In addition, compared with wild-type EZH2, USP36 displayed decreased interaction with EZH2 K222R (Figure 5F). Thus, our data support the hypothesis that USP36 is the deubiquitinase that is responsible for EZH2 K222 deubiquitination.
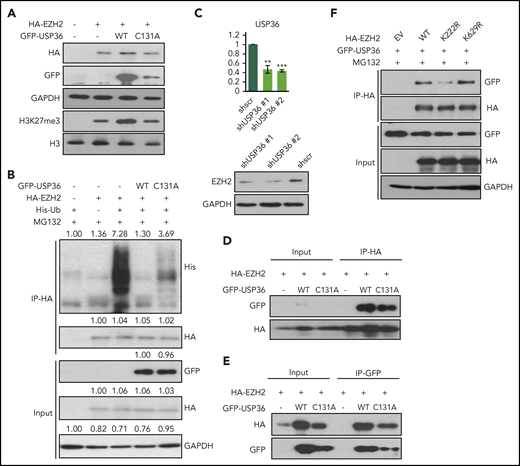
USP36 mediates EZH2 K222 deubiquitination and stabilizes EZH2. (A) USP36 overexpression led to the stabilization of exogenously overexpressed EZH2, which could be rescued by USP36 C131A mutant transfection in HEK293T cell. Cells were harvested for immunoblots 48 hours after transfection. (B) Overexpression of USP36 led to deubiquitination of EZH2 in HEK293T cells, and this is partially rescued by USP36 C131A mutant transfection. MG132 was given at 5 µM for 6 hours of treatment 48 hours after transfection. Densitometry analysis was used to quantify average changes in 3 individual experiments. (C) Knock-down of USP36 led to a decrease of EZH2 protein in KHYG-1 cells. The mRNA level of USP36 was indicated. Data are shown as mean ± SD, N = 3. *P < .05; **P < .01; ***P < .001. Cells were harvested for RNA extraction and immunoblots 24 hours after transfection. (D-E) Interaction between exogenously overexpressed EZH2 and USP36 wild-type or C131A mutant. Cells were harvested for co-IP 48 hours after transfection. (F) Change of interaction between USP36 and EZH2 wild-type or its mutants in HEK293T cells. Cells were harvested for co-IP 48 hours after transfection. All immunoblots were performed in at least 3 individual experiments; representative images are shown.
USP36 mediates EZH2 K222 deubiquitination and stabilizes EZH2. (A) USP36 overexpression led to the stabilization of exogenously overexpressed EZH2, which could be rescued by USP36 C131A mutant transfection in HEK293T cell. Cells were harvested for immunoblots 48 hours after transfection. (B) Overexpression of USP36 led to deubiquitination of EZH2 in HEK293T cells, and this is partially rescued by USP36 C131A mutant transfection. MG132 was given at 5 µM for 6 hours of treatment 48 hours after transfection. Densitometry analysis was used to quantify average changes in 3 individual experiments. (C) Knock-down of USP36 led to a decrease of EZH2 protein in KHYG-1 cells. The mRNA level of USP36 was indicated. Data are shown as mean ± SD, N = 3. *P < .05; **P < .01; ***P < .001. Cells were harvested for RNA extraction and immunoblots 24 hours after transfection. (D-E) Interaction between exogenously overexpressed EZH2 and USP36 wild-type or C131A mutant. Cells were harvested for co-IP 48 hours after transfection. (F) Change of interaction between USP36 and EZH2 wild-type or its mutants in HEK293T cells. Cells were harvested for co-IP 48 hours after transfection. All immunoblots were performed in at least 3 individual experiments; representative images are shown.
Because USP36 has previously been described as a nucleolar deubiquitinase, it is probable that nucleolus is the main location for USP36 to deubiquitinate EZH2. As illustrated, interaction between USP36 and EZH2 was found in the nucleolus but not in the nucleoplasm (supplemental Figure 5A-B). Furthermore, ectopic expression of USP36 decreased EZH2 ubiquitination in the nucleolus (supplemental Figure 5C). Taken together, these data suggest that nucleolus may be the major site of USP36-mediated EZH2 deubiquitination.
Stabilization of EZH2 modulates chemo-sensitivity to bortezomib treatment
As the first-generation proteasome inhibitor, bortezomib has already been clinically approved for the treatment of myeloma and mantle cell lymphoma. In recent clinical trials, bortezomib was used for NKTL treatment in combination of other chemotherapy agents or regimens, such as panobinostat22 or cyclophosphamide, hydroxydaunorubicin, oncovin, and prednisone (CHOP)23,24 regimen, and satisfactory synergistic effects were observed in NKTL patients. Notably, EZH2 deregulation was shown to contribute to loss of sensitivity in Bortezomib treatment in multiple myeloma through the CDK-RB-E2F axis.25,26 Therefore, we speculated that changes of EZH2 stability may also link to bortezomib sensitivity in NKTL.
In NKTL cells, overexpression of EZH2 resulted in attenuated sensitivity to bortezomib treatment (Figure 6A-B). And in contrast, when EZH2 was knocked down, sensitized response to bortezomib treatment was seen (Figure 6C-D). As anticipated, MELK overexpression diminished bortezomib sensitivity in NKTL cells compared with vector, and this could be rescued by MELK T167A (attenuated enzymatic activity) transfection (Figure 6E-F). In line with these results, knock-down or chemical inhibition of MELK sensitized NKTL cells to bortezomib (Figure 6G-H; supplemental Figure 6), and synergistic effects between bortezomib and MELK inhibitor OTSSP167 were seen. Notably, EZH2 S220A mutant sensitized the NKTL cells to bortezomib treatment compared with EZH2 wild-type (Figure 6I-J). In comparison, manipulation of MELK expression in NKTL cells could not affect sensitivity to a different proteasome inhibitor, carfilzomib, which has not yet been approved for any NKTL trial (supplemental Figure 7). Collectively, these data suggest that MELK, partly through the regulation of EZH2 ubiquitination, may to some extent attenuate sensitivity to bortezomib treatment in NKTL.
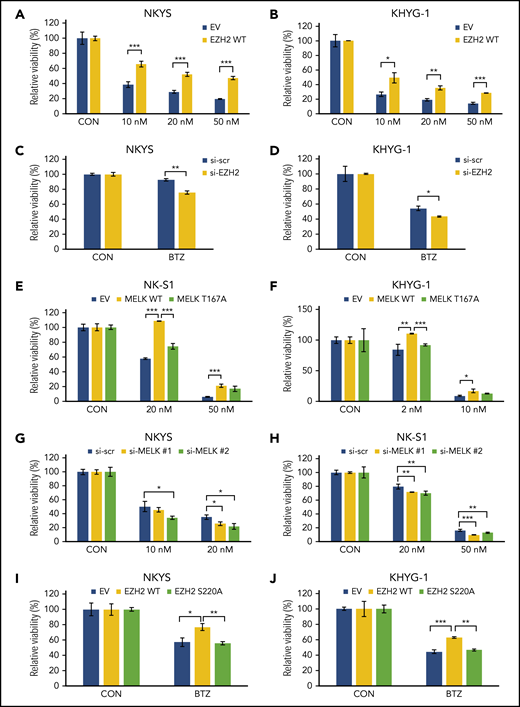
EZH2 loss of ubiquitination mediates bortezomib-sensitivity for NKTL cell lines. (A-B) Relative cell viability for treatment with bortezomib in (A) NKYS and (B) KHYG-1 cells overexpressing EZH2 wild-type. (C-D) Relative cell viability for treatment with bortezomib on (C) NKYS (5 nM) and (D) KHYG-1 (10 nM) cells upon EZH2 knock-down using siRNA. (E-F) Relative cell viability for treatment with bortezomib on (E) NK-S1 and (F) KHYG-1 cells overexpressing MELK wild-type or MELK T167A mutant. (G-H) Relative cell viability for treatment with bortezomib in (G) NKYS and (H) NK-S1 cells upon MELK knock-down. (I-J) Relative cell viability for treatment with 5 nM bortezomib in (I) NKYS and (J) KHYG-1 cells overexpressing EZH2 wild-type or EZH2 S220A mutant. Twenty hours of bortezomib treatment was used in all of these experiments. Cell survival was measured using CellTiter-Glo reagent. For all siRNA knock-down, bortezomib was added 48 hours after transfection. For all plasmid transfections, bortezomib was added 20 hours after transfection, and puromycin selection were used (1 µg/mL) along with bortezomib treatment. Data are shown as mean ± SD, N ≥ 3. *P < .05; **P < .01; ***P < .001.
EZH2 loss of ubiquitination mediates bortezomib-sensitivity for NKTL cell lines. (A-B) Relative cell viability for treatment with bortezomib in (A) NKYS and (B) KHYG-1 cells overexpressing EZH2 wild-type. (C-D) Relative cell viability for treatment with bortezomib on (C) NKYS (5 nM) and (D) KHYG-1 (10 nM) cells upon EZH2 knock-down using siRNA. (E-F) Relative cell viability for treatment with bortezomib on (E) NK-S1 and (F) KHYG-1 cells overexpressing MELK wild-type or MELK T167A mutant. (G-H) Relative cell viability for treatment with bortezomib in (G) NKYS and (H) NK-S1 cells upon MELK knock-down. (I-J) Relative cell viability for treatment with 5 nM bortezomib in (I) NKYS and (J) KHYG-1 cells overexpressing EZH2 wild-type or EZH2 S220A mutant. Twenty hours of bortezomib treatment was used in all of these experiments. Cell survival was measured using CellTiter-Glo reagent. For all siRNA knock-down, bortezomib was added 48 hours after transfection. For all plasmid transfections, bortezomib was added 20 hours after transfection, and puromycin selection were used (1 µg/mL) along with bortezomib treatment. Data are shown as mean ± SD, N ≥ 3. *P < .05; **P < .01; ***P < .001.
Discussion
In NKTL, the mechanism of EZH2 overexpression is not thoroughly defined and all the commercialized small-molecule or peptide inhibitors do not target the enzymatically independent oncogenic activity of EZH2, it is important to better understand the biological turnover of EZH2 protein in this malignancy. In this study, we demonstrate that the serine/threonine kinase MELK directly phosphorylates EZH2 at S220, resulting in a corresponding downregulation of EZH2 ubiquitination at K222. Our findings identify a previous undescribed mechanism of EZH2 protein stabilization mediated through MELK phosphorylation, and EZH2-associated loss of sensitivity to bortezomib may be further exacerbated by MELK overexpression in NKTL through the stabilization of EZH2.
In a majority of contexts, EZH2’s tumorigenic effect is mediated through its enzymatic function of catalyzing H3K27 trimethylation. In these cases, oncogenic EZH2 can be suppressed by small-molecule enzymatic inhibitors, whereas in a few cancers, including NKTL, the tumorigenic role of overexpressed EZH2 is independent of its catalytic function and the PRC2 complex.1,11,27 Accordingly, enzymatic inhibition or PRC2 disruption is ineffective against EZH2-mediated oncogenesis in NKTL. In addition, we have demonstrated previously that JAK3 functionally switches the role of EZH2 to a gene activator and mediates tumorigenicity through EZH2 Y244 phosphorylation in NKTL.14 JAK3 inhibition effectively blocks EZH2 oncogenesis by disrupting oncogenic phosphorylation of EZH2,14 yet the basal level of EZH2 protein remains intact. And the possibility that the Y244-unphosphorylated EZH2 still favors NKTL cells cannot be ruled out.
Besides the direct conferment of survival advantages, EZH2 overexpression also plays a role in chemo-resistance in a variety of cancers. Bortezomib is clinically approved for the treatment of multiple myeloma and mantle cell lymphoma. It demonstrated therapeutic effects for NKTL treatment in clinical trials in combination with other chemotherapy regimens.22 In multiple myeloma, EZH2 deregulation exposes the myeloma cells to a loss of sensitivity to bortezomib treatment through the CDK-RB-E2F axis.25,26 In our study, we find that stabilization of EZH2 by MELK results in enhanced resistance to bortezomib treatment in NKTL, and this relies on the S220-specific phosphorylation of EZH2. Enzymatic defection of MELK or phospho-dead mutation of EZH2 may compromise EZH2-mediated bortezomib resistance. This might be linked to the reversible effect28 of bortezomib on proteasome as sensitivity to the advanced and irreversible28 proteasome inhibitor carfilzomib, which might also mediate the CDK pathway,26 was not affected by MELK expression in NKTL cells. The mechanisms underscoring these differences will need to be examined in future studies.
EZH2 phosphorylation is one of the key posttranslational events that modulates the activity and stability of EZH2 and exerts various biological influences. S21 phosphorylation of EZH2 by AKT, which has been found in multiple cellular context, impairs its histone methyltransferase activity and is functionally important for EZH2-associated signaling pathways.29 It is critical for EZH2 to mediate carcinogenesis or chemo-sensitivity in glioma,30 hepatocellular carcinoma31 and multiple myeloma32 through different mechanisms. This phosphorylation is also indispensable for the PRC2-independent role of EZH2 in castration-resistant prostate cancer.27 Moreover, CDK-mediated EZH2 phosphorylation at several sites inhibits H3K27 trimethylation and regulates embryonic stem cell divisions.33-35 As mentioned previously, our previous study reveals that EZH2 Y244 phosphorylation by JAK3 converts EZH2 from an epigenetic silencer to a transcriptional activator in NKTL.14 In this study, we have identified MELK kinase, which co-overexpresses with EZH2, phosphorylates EZH2 at S220, and promotes USP36-mediated K222 deubiquitination in the same malignancy. These 2 phosphorylation sites, though both identified in NKTL, are functionally and spatially different. The Y244 phosphorylation renders EZH2 to get rid of the PRC2 complex and bind to POL II, therefore switching to an activating effector. The basal protein level of EZH2 is not affected. In comparison, S220 phosphorylation associates with proteasomal degradation of EZH2. On the other hand, as POL II mainly facilitates transcription in the nucleoplasm, by binding to POL II the Y244-phosphorylated EZH2 coactivates transcription within the same region, whereas USP36 mainly locates in the nucleolus; thus, the localization of EZH2 K222 deubiquitination may be within the nucleolus.
The MELK-EZH2 axis has been reported before. In glioma stem cells, MELK could phosphorylate and activate FOXM1, which then binds and transactivates EZH2 expression, resulting in favored survival and radioresistance.12,17 Colocation and interaction between MELK and EZH2 were also observed in medulloblastoma.18 Our study uncovers a new mechanistic link between MELK and EZH2 that MELK phosphorylates EZH2 and directs EZH2 to escape proteasomal degradation. Unlike glioma, FOXM1 is not an essential mediator of EZH2 in NKTL malignancy.
Several previous studies have revealed that EZH2 protein stability could be modulated by the ubiquitin-proteasome system. A few E3 ligases of EZH2, such as Praja1,36 TRAF6,9 and β-TrCP,37 have been identified and functionally clarified in these years. Very recently, ZRANB1 was reported as a deubiquitinase of EZH2 in triple-negative breast cancer.38 In NKTL, this study has identified USP36 as the deubiquitinase that mediates EZH2 K222 deubiquitination. As a nucleolar deubiquitinase, the regulation of USP36 on EZH2 hints at the nucleolus localization of EZH2. This correlates to the findings that EZH2 interacts with a variety of ribonucleoproteins, ribosome components, and other nucleolar proteins which have been identified by several high throughput affinity-captured MS analysis.39-41
In summary, we demonstrate in this study for the first time that in NKTL, overexpressed MELK specifically phosphorylates EZH2 at S220 and mediates stabilization of EZH2 with the assistance of USP36.
The publication costs of this article were defrayed in part by page charge payment. Therefore, and solely to indicate this fact, this article is hereby marked “advertisement” in accordance with 18 USC section 1734.
The gene expression profiling data regarding MELK expression have been deposited in the Gene Expression Omnibus database (accession number GSE90784).
The MS data have been deposited to the ProteomeXchange Consortium via PRIDE (PXD015008).
The online version of this article contains a data supplement.
Acknowledgments
The authors thank Yu Qiang (Genome Institute of Singapore) for pcDNA hEZH2 and pMN-FOXM1 plasmids; Edward Chow for carfilzomib; Adina Nee for cell pellets of normal NK, SNK1, SNK6, HANK-1, and NK-92; Brittney Foo for preparing materials for some immunoblots; and Daniel G. Tenen, Takaomi Sanda, and Motomi Osato for critical comments.
This research was supported by the National Research Foundation Singapore and the Singapore Ministry of Education under the Research Centres of Excellence initiative as well as the RNA Biology Centre at the Cancer Science Institute of Singapore, National University of Singapore (MOE2014-T3-1-006). W-J.C. was also supported by National Medical Research Council Singapore Translational Research Investigatorship.
Authorship
Contribution: W.-J.C., J.Y., and B. Li conceived the study and designed experiments; B. Li, T.P., B. Lin, and S.F. performed experiments; B. Li, D.K., L.W., B.-C.G., T.-H.C., and S.-B.N. analyzed and interpreted data; S.-B.N. provided gene expression data on natural killer/T-cell lymphoma clinical samples; P.J.A.E. provided essential constructs or short hairpin RNAs for USP36, USP48, and ubiquitin; and B. Li, W.-J.C., P.J.A.E., D.K., T.P., S.-B.N., and N.M. wrote the manuscript.
Conflict-of-interest disclosure: The authors declare no competing financial interests.
Correspondence: Wee-Joo Chng, Cancer Science Institute of Singapore, National University of Singapore, NUHS Tower Block #7, 1E Kent Ridge Road, 119228, Singapore; e-mail: mdccwj@nus.edu.sg.

This feature is available to Subscribers Only
Sign In or Create an Account Close Modal