

Key Points
Mutations in the transcription factor IKZF5 cause autosomal dominant thrombocytopenia and a paucity of α granules.
Although IKZF5 is expressed across hematopoietic lineages, misregulation in IKZF5 cases is restricted to the megakaryocytic lineage.

Abstract
To identify novel causes of hereditary thrombocytopenia, we performed a genetic association analysis of whole-genome sequencing data from 13 037 individuals enrolled in the National Institute for Health Research (NIHR) BioResource, including 233 cases with isolated thrombocytopenia. We found an association between rare variants in the transcription factor-encoding gene IKZF5 and thrombocytopenia. We report 5 causal missense variants in or near IKZF5 zinc fingers, of which 2 occurred de novo and 3 co-segregated in 3 pedigrees. A canonical DNA-zinc finger binding model predicts that 3 of the variants alter DNA recognition. Expression studies showed that chromatin binding was disrupted in mutant compared with wild-type IKZF5, and electron microscopy revealed a reduced quantity of α granules in normally sized platelets. Proplatelet formation was reduced in megakaryocytes from 7 cases relative to 6 controls. Comparison of RNA-sequencing data from platelets, monocytes, neutrophils, and CD4+ T cells from 3 cases and 14 healthy controls showed 1194 differentially expressed genes in platelets but only 4 differentially expressed genes in each of the other blood cell types. In conclusion, IKZF5 is a novel transcriptional regulator of megakaryopoiesis and the eighth transcription factor associated with dominant thrombocytopenia in humans.
Introduction
More than 30 genes have been implicated in hereditary thrombocytopenia,1 of which 7 genes encode lineage-specific transcription factors (TFs).2 Variants in 2 of these genes (RUNX1 and ETV6) increase the risk of leukemia,3,4 whereas others are associated with bone marrow failure (HOXA11 and MECOM), red blood cell defects (GATA1 and GFI1B), or syndromic disorders (FLI1).5-8
IKZF5 (Pegasus) is 1 of the 5 members of the Ikaros family of TF genes. IKZF1 through 3 (Ikaros, Helios, and Aiolos), have established roles in lymphocyte development,9 with germline mutations in IKZF1 implicated in common variable immunodeficiency and somatic variants in both IKZF1 and IKZF3 associated with hematological malignancies.10-12 In contrast, the functions of IKZF4 (Eos) and IKZF5, which are both widely expressed in hematopoietic cells,13 have remained unknown. IKZF5 encodes a DNA-binding factor that recognizes elements containing the core sequence GNNTGTNG via its 3 N-terminal zinc fingers (Znfs), whereas the C-terminal Znfs control self-association of Ikaros proteins. IKZF5 is highly homologous to the other Ikaros proteins except at the N terminus, where it contains 3 rather than 4 Znfs and has distinct DNA-binding sites.14,15 Here, we present genetic and functional evidence demonstrating that rare missense variants in IKZF5 cause thrombocytopenia.
Methods
Enrollment, research ethics, and consent
Enrollment of participants in the National Institute for Health Research (NIHR) BioResource–Rare Diseases occurred between December 2012 and March 2017. All participants provided written informed consent either under the East of England Cambridge South national research ethics committee reference 13/EE/0325 or alternate research ethics committee-approved studies for UK patients; obtaining consent for overseas samples was the responsibility of the respective principal investigator at the enrolling hospitals following oversight of the consent form for their study and a material transfer agreement.16
Clinical and laboratory phenotype data
We collected clinical data from probands and pedigree members and coded them with Human Phenotype Ontology terms as described previously.17 Individuals with isolated thrombocytopenia were defined as those who had a Human Phenotype Ontology term “thrombocytopenia” or a platelet count (PLT) <130 × 109/L and were deemed nonsyndromic (ie, did not have any severe abnormalities in other organ systems).
DNA sequencing and variant calling
DNA extraction, library preparation for whole-genome sequencing (WGS) and whole-exome sequencing (WES), variant calling, and variant annotation were performed as described previously.16,17
Variant confirmation and cosegregation studies
Variants called by WGS were confirmed and tested for in pedigree members by polymerase chain reaction (PCR) and Sanger sequencing. Primers were designed with Primer3Plus (https://primer3plus.com/cgi-bin/dev/primer3plus.cgi). Routine PCR was performed using MyTaq HS Mix (BIOLINE BIO-25045), a 2× Hot-start mix. Primers were used at a final concentration of 0.5 µM in 20-µL reactions. Thermal cycling using a Touchdown protocol,18 with GeneAmp PCR System 9700 (Applied Biosystems, ThermoFisher). The Cycling protocol was an initial denature step of 3 minutes at 95°C, then 15 cycles of 95°C for 30 seconds, 70°C 1-minute touchdown (1°C/cycle), and 72°C for 30 seconds. The final cycling phase was 25 cycles of 95°C for 30 seconds, 55°C for 1 minute, and 72°C for 30 seconds, with a final extension of 72°C for 5 minutes. The PCR products were checked by agarose gel electrophoresis, column purified using QIAquick PCR Purification kit (QIAGEN catalog no. 28106) and eluted with 30 µL nuclease free water. The amplicons were quantified with QUBIT dsDNA BR assay kit (Life Technologies Ltd [Invitrogen Division] catalog no. Q32853). Purified, quantified amplicons were sent to Source Bioscience for Sanger sequencing, with the same primers that were used to amplify the target. The primers used were 5′-TCCAATGAATGGACAAGCAA-3′ and 5′-TCGAGAGCTGATTCAAAGGG-3′, for variants at GRCh37 positions 10:124754093, 10:124754138, and 10:124754155, targeting a 530-bp amplicon. Variants called by WES were confirmed and tested for in pedigree members following a similar approach. The primers used were 5′-TTTTGTGAGAAGTGAACTGTC-3′ and 5′-AGGGCATCACTTCTGACCTGT-3′, for variants at GRCh37 positions 10:124755560 and 10:124755540, targeting a 491-bp amplicon.
Genetic association analysis
We applied BeviMed19 to compute a probability of association between each gene and case/control status, defined by presence or absence of isolated thrombocytopenia, among unrelated individuals, as described previously.16 In general, we excluded somatic short variants/indels with an allele count in any GnomAD population in excess of what would be expected given a true population minor allele frequency of 1/10 000 (under a dominant model) or 1/1000 (under a recessive model).16 Large deletions with an internal allele count among unrelated Europeans or non-Europeans in excess of what would be expected given a population allele frequency of 1/200 were also excluded.16 To boost power, when assessing the evidence for genetic association with thrombocytopenia for a particular gene, cases with explanatory variants in other genes were treated as controls. Thus, the numbers of cases and controls varied slightly by gene, depending on whether the gene being assessed contained any cases carrying known pathogenic variants. Variant selection on the basis of allele frequency and predicted consequence was performed as described previously.16 BeviMed compares the statistical support for a baseline model in which disease risk is independent of the genotypes with various association models in which disease risk depends on the allele configuration at the given rare variant sites, a latent partition of variants into pathogenic and benign groups, and a Mendelian mode of inheritance. The association models are fitted to different subsets of variants in a given locus corresponding to different predicted consequences, which imposes a prior correlation structure on the pathogenicity of the variants that reflects competing potential disease mechanisms.
Electron microscopy
We performed transmission electron microscopy (EM) analysis of platelets as described previously.20 The platelet surface area (in square micrometers) and the number of α granules per unit area (in 1/μm2) were measured blinded to case/control status as done previously for GATA1- and NBEAL2-related granule defects.21 Whole mount EM analysis to quantify dense granules was performed as described previously.22
Megakaryocyte differentiation assays
CD34+ hematopoietic stem cells (HSCs) were isolated by magnetic cell sorting (Miltenyi Biotec, Bergisch Gladback, Germany) from peripheral blood. The recovered (differentiation day 0) CD34+ HSCs were cultured in StemSpan SFEM medium with StemSpan CC100 ensuring expansion of HSC for 3 days (Stem Cell Technologies, Vancouver, Canada). Liquid megakaryocyte (MK) cultures were obtained by incubation with 50 ng/mL thrombopoietin, 25 ng/mL stem cell factor, and 10 ng/mL interleukin 1β (Peprotech, Rocky Hill, NJ). MKs were analyzed by flow cytometry on total differentiation day 10 for CD41 and CD42 markers. Proplatelet formation (PPF) was quantified after immunostaining of the cytoskeletal protein F-actin with phalloidin-rhodamine (Sigma, St Louis, MO) and α granule marker von Willebrand factor (VWF; A0082; Dako). For immunostaining, MKs were seeded for 4 hours on fibrinogen-coated coverslips to allow PPF; stained cells were photographed at 63× magnification with a confocal microscope (AxioObserver Z1; Zeiss, Heidelberg, Germany). PPF of MKs was assessed by random imaging of 40 MKs per condition.
Structural modeling
We modeled the functional consequences of the IKZF5 mutations using the structural coordinates of Znf 4 of CCCTC-binding factor (CTCF), the closest homolog of known structure to IKZF5 (PDB entry 5kkq). We did a BLAST search against the UniRef90 database23 to find unique relatives for the sequence covering Znfs 1 to 3 of IKZF5 (residues 84-161). Structural consequences of the mutations of this region of IKZF5 were evaluated in the context of the closest homolog of known structure. We used CCP4mg software24 to visualize the 3-dimensional structure of IKZF5 domains
HEK293 expression and localization studies
We cloned wild-type (WT) and mutant IKZF5 in the pSecTag2/HygroA vector (Life Sciences) to transfect HEK293 cells. We prepared protein extracts 48 hours after transfection using the subcellular protein fractionation kit for cultured cells (Thermo Scientific) to isolate chromatin-bound proteins. We performed western blotting using the following antibodies: rabbit polyclonal anti-IKZF5 (Atlas), rabbit monoclonal anti-glyceraldehyde-3-phosphate dehydrogenase (clone 14C10; Cell Signaling) and goat polyclonal anti-Histone H3 or HIST3H3 (clone C16; Santa Cruz). We performed staining with the ECL detection reagent (Life Technologies) and imaged chemiluminescent blots with the ChemiDoc MP imager and the ImageLab software, version 4.1 (Bio-Rad). Transfected cells were analyzed with a confocal microscope (AxioObserver.Z1) and a structured illumination microscope (Elyra S.1; Zeiss) was used for z-stack images. Images were analyzed with ZEN Black (Zeiss).
RNA-sequencing study
For each studied individual, we separated peripheral blood mononuclear cells by gradient centrifugation (Percoll 1.078 g/mL) and isolated neutrophils from the pellet after double red blood cell lysis. Peripheral blood mononuclear cells were further separated into monocytes by CD14+ selection (Miltenyi) and total CD4 (Stem Cell Technologies) from the CD14 column flow through. Platelets were isolated from platelet-rich plasma after leukocyte (CD45+) depletion as described previously.25 All protocols used are available at http://www.blueprint-epigenome.eu/. Purified cells were resuspended in Trizol, from which RNA was extracted following the manufacturer's instructions. For each cell type, reverse transcription and preparation of a high-throughput sequencing library was performed using the Riboerase Kapa stranded RNA kit following the manufacturer's instructions. We generated 150-bp paired-end reads on an Illumina Hiseq4000. We used TrimGalore, version 0.3.7 (http://www.bioinformatics.babraham.ac.uk/projects/trim_galore/), with parameters “-q 15 -s 3 -length 30 -e 0.05” to trim PCR and sequencing adapters. Trimmed reads were aligned to the Ensembl version 8026 human transcriptome with Bowtie 1.0.127 using the parameters “-a -best -strata -S -m 100 -X 500 -chunkmbs 256 -nofw -fr.” We used MMSEQ (version 1.0.8a)28 with default parameters to quantify gene expression. We inferred differential expression using MMDIFF.29 Genes with a posterior probability (PP) of differentially expression >0.8 were considered differentially expressed. We performed Reactome pathway enrichment analysis using the compareCluster and enrichPathway functions from the ReactomePA Bioconductor R package.30
Results
Genetic analysis highlights IKZF5 as a candidate gene for isolated thrombocytopenia
Using the BeviMed method for genetic association,19 for each gene we compared the genotypes at rare variant sites of up to 105 unrelated unexplained individuals having isolated thrombocytopenia (cases) with the genotypes of up to 10 053 unrelated individuals unaffected by thrombocytopenia or with thrombocytopenia caused by variants in other genes (controls) (see “Methods”). Of 33 814 genes assessed, 16 yielded a PP of association >0.4, of which 13 are established genes implicated in thrombocytopenia (Figure 1A). The associations for CSF3 and ELMOD1 were likely not causal because their high PPs depended on variants absent from affected relatives of thrombocytopenic carriers being pathogenic. The BeviMed association between thrombocytopenia and IKZF5 was driven by 4 rare missense variants. We next reviewed an in-house dataset of 534 samples analyzed by WES from other individuals enrolled in the NIHR BioResource, and identified 2 additional thrombocytopenic cases carrying 2 different rare missense variants in IKZF5. All 6 missense variants were absent from gnomAD and 5 variants, encoding Y89C, R96W, G134E, C140R, and H155Y, were at evolutionarily conserved residues located in or near the N-terminal Znfs and were thus selected for further detailed analysis, while a sixth missense variant, affecting residue 200 (S200G), was expected to be benign because it was located far from Znfs and outside evolutionarily conserved regions (Figure 1B).

Rare missense variants in IKZF5 are associated with thrombocytopenia. (A) BeviMed was applied gene by gene to infer associations between the genotypes of filtered rare variants and a case/control grouping defined by isolated thrombocytopenia. The posterior probabilities for genetic association inferred by BeviMed exceeding 0.4 are shown. The dots representing posterior probabilities for genes previously known to be implicated in hereditary thrombocytopenia are highlighted in blue. Genes not previously associated with thrombocytopenia are in gray. (B) Evolutionary conservation scores with respect to 66 protein sequences obtained using multiple sequence alignment within ConSurf,31 and the corresponding 95% confidence intervals, for amino acids (AAs) 1 through 419 of IKZF5, normalized to have a mean of 0 and standard deviation of 1. The filled/empty points indicate presence/absence of missense variants in GnomAD altering the AAs. Only 3 of the 141 456 individuals in GnomAD harbor a missense variant affecting a residue in the N-terminal Znfs with a normalized conservation score <−1 (AAs 102, 111, and 123). The blue boxes indicate the locations of the Znfs. The variants in cases with thrombocytopenia affecting the 5 conserved AAs have been followed up in cosegregation studies, while S200G, which affects a non-conserved AA, has not been followed up. (C) Sex-stratified histograms of PLT obtained using a Sysmex hematology analyzer from 48 345 blood donors from the Efficiency and Safety of Varying the Frequency of Whole Blood Donation study32 after adjustment for technical artifacts. The red arrows superimposed on the histograms indicate the sex of and values for cases carrying 1 of the 5 missense rare variants. The green arrows indicate the sex of and values for relatives homozygous for the corresponding WT allele. Individual C II.3, marked with an asterisk, had thrombocytopenia as a child (with an unknown PLT) and was subsequently splenectomized, likely explaining why his PLT increased to 253 × 109/L as an adult. (D) The 5 pedigrees recalled for cosegregation. Male (square) and female (circle) individuals are shown in black or white depending on whether they are affected or unaffected, respectively, whereas individuals with an unknown PLT are shown in gray. Genotyping results obtained by WGS (G), WES (E), or Sanger sequencing (S) are shown in terms of their predicted amino acid substitutions underneath the genotyped individuals. Crl, control; WT, homozygous for the reference allele. C III.4 carries the G134E variant but has a PLT of 184.
Rare missense variants in IKZF5 are associated with thrombocytopenia. (A) BeviMed was applied gene by gene to infer associations between the genotypes of filtered rare variants and a case/control grouping defined by isolated thrombocytopenia. The posterior probabilities for genetic association inferred by BeviMed exceeding 0.4 are shown. The dots representing posterior probabilities for genes previously known to be implicated in hereditary thrombocytopenia are highlighted in blue. Genes not previously associated with thrombocytopenia are in gray. (B) Evolutionary conservation scores with respect to 66 protein sequences obtained using multiple sequence alignment within ConSurf,31 and the corresponding 95% confidence intervals, for amino acids (AAs) 1 through 419 of IKZF5, normalized to have a mean of 0 and standard deviation of 1. The filled/empty points indicate presence/absence of missense variants in GnomAD altering the AAs. Only 3 of the 141 456 individuals in GnomAD harbor a missense variant affecting a residue in the N-terminal Znfs with a normalized conservation score <−1 (AAs 102, 111, and 123). The blue boxes indicate the locations of the Znfs. The variants in cases with thrombocytopenia affecting the 5 conserved AAs have been followed up in cosegregation studies, while S200G, which affects a non-conserved AA, has not been followed up. (C) Sex-stratified histograms of PLT obtained using a Sysmex hematology analyzer from 48 345 blood donors from the Efficiency and Safety of Varying the Frequency of Whole Blood Donation study32 after adjustment for technical artifacts. The red arrows superimposed on the histograms indicate the sex of and values for cases carrying 1 of the 5 missense rare variants. The green arrows indicate the sex of and values for relatives homozygous for the corresponding WT allele. Individual C II.3, marked with an asterisk, had thrombocytopenia as a child (with an unknown PLT) and was subsequently splenectomized, likely explaining why his PLT increased to 253 × 109/L as an adult. (D) The 5 pedigrees recalled for cosegregation. Male (square) and female (circle) individuals are shown in black or white depending on whether they are affected or unaffected, respectively, whereas individuals with an unknown PLT are shown in gray. Genotyping results obtained by WGS (G), WES (E), or Sanger sequencing (S) are shown in terms of their predicted amino acid substitutions underneath the genotyped individuals. Crl, control; WT, homozygous for the reference allele. C III.4 carries the G134E variant but has a PLT of 184.
Cosegregation studies showed that H155Y and C140R, in pedigrees A and B, respectively (Figure 1D), occurred de novo, with the latter appearing in monozygotic twins. Cosegregation studies of G134E, Y89C, and R96W, in pedigrees C, D, and E, respectively, identified 10 additional relatives carrying mutant alleles and 2 unaffected relatives homozygous for the WT allele. Only 2 of the 10 individuals carrying the mutant allele had PLT >150 × 109/L: a father and son in pedigree C. Case C II.3 was a 53-year-old male with a PLT in adulthood of 253 × 109/L who had undergone a splenectomy as a child for treatment of thrombocytopenia. One of his sons (C III.1) had a PLT level of 183 × 109/L, which is in the 11.6th percentile of the PLT distribution for healthy males32 (Figure 1C; Table 1). Unfortunately, he was not available to obtain a second PLT measurement or for more detailed phenotyping. All other individuals who carried the mutant IKZF5 allele across the 5 pedigrees had PLT <150 × 109/L, with a normal platelet size (Table 1). Overall, 8 of 16 carriers (ie, 50%) had a PLT <100 × 109/L and none had a PLT <50 × 109/L. Because other members of the Ikaros family of Znf proteins are critical for lymphoid development and are associated with immune dysregulation and leukemia in humans, we specifically inquired about these features in the 5 pedigrees. No leukemias were reported in any of the pedigrees and there were no signs of immunodeficiency, such as recurrent infections. Immunoglobulin levels were normal in all cases for whom they were available (Table 1).
Information on pedigree members
| Pedigree | ID | Sex | Genotype | Sequencing | TCP | PLT | MPV | BD | Blood film analysis | α granule quantity by EM | Immunoglobulins |
|---|---|---|---|---|---|---|---|---|---|---|---|
| A | I.1 | M | WT | Sanger | N | 211 | 11.4 | ||||
| A | I.2 | F | WT | Sanger | N | 276 | 10.5 | ||||
| A | II.1 | M | H155Y het* | WGS | Y | 76 | 10.2 | None | Reduced | Normal | |
| A | II.2 | M | WT | Sanger | N | 193 | 10.8 | ||||
| B | I.1 | M | WT | Sanger | N | 173 | |||||
| B | I.2 | F | WT | Sanger | N | 301 | |||||
| B | II.1 | F | C140R het* | WGS | Y | 78 | 10.4 | None | TCP, all else normal | Reduced | Normal |
| B | II.2 | F | C140R het* | WGS | Y | 83 | 10.2 | None | TCP, all else normal | Reduced | Normal |
| B | II.3 | F | WT | Sanger | N | 257 | 10.9 | Normal | |||
| C | II.3 | M | G134E het | WGS | N | 253† | 12.5 | ||||
| C | II.4 | F | WT | Sanger | N | 327 | 11.4 | ||||
| C | III.1 | M | G134E het | Sanger | Y | 128 | 9.4 | ||||
| C | III.3 | F | G134E het | WGS | Y | 82 | 8.6 | None | TCP, all else normal | ||
| C | III.4 | M | G134E het | Sanger | N | 184 | 11.1 | ||||
| C | IV.1 | M | G134E het | Sanger | Y | 82 | 8.8 | ||||
| D | I.1 | M | N | ||||||||
| D | I.2 | F | Y | ||||||||
| D | II.2 | F | Y | 110 | 8.2 | ||||||
| D | II.3 | F | Y89C het | Sanger | Y | 97 | 8.9 | Normal | |||
| D | III.1 | M | N | 195 | 9 | ||||||
| D | III.2 | F | Y89C het | WES | Y | 70 | 8.3 | ||||
| E | I.2 | F | Y | ||||||||
| E | II.1 | M | N | 255 | |||||||
| E | II.2 | M | Y | 155 | |||||||
| E | II.3 | M | R96W het | WES | Y | 107 | 9.5 | None | Normal | Reduced | Normal |
| E | II.4 | F | WT | Sanger | N | 220 | 9 | ||||
| E | II.6 | M | R96W het | Sanger | Y | 111 | 10.4 | None | Normal | Reduced | Normal |
| E | II.8 | F | R96W het | Sanger | Y | 104 | 11 | Mild | Normal | Reduced | Normal |
| E | III.1 | F | R96W het | Sanger | Y | 96 | 8.9 | None | |||
| E | III.2 | M | R96W het | Sanger | Y | 110 | 9.4 | None | |||
| E | III.3 | F | Y | 127 | 11 | ||||||
| E | III.4 | M | Y | 135 | 8.8 | ||||||
| E | III.5 | F | Y | 106 | 12 | ||||||
| E | III.6 | M | Y | 140 | 11.9 | ||||||
| E | III.7 | F | R96W het | Sanger | Y | 117 | 11.4 | Mild | Normal | Reduced | Normal |
| E | III.9 | M | N | 275 | |||||||
| E | IV.1 | F | Y | 110 |
| Pedigree | ID | Sex | Genotype | Sequencing | TCP | PLT | MPV | BD | Blood film analysis | α granule quantity by EM | Immunoglobulins |
|---|---|---|---|---|---|---|---|---|---|---|---|
| A | I.1 | M | WT | Sanger | N | 211 | 11.4 | ||||
| A | I.2 | F | WT | Sanger | N | 276 | 10.5 | ||||
| A | II.1 | M | H155Y het* | WGS | Y | 76 | 10.2 | None | Reduced | Normal | |
| A | II.2 | M | WT | Sanger | N | 193 | 10.8 | ||||
| B | I.1 | M | WT | Sanger | N | 173 | |||||
| B | I.2 | F | WT | Sanger | N | 301 | |||||
| B | II.1 | F | C140R het* | WGS | Y | 78 | 10.4 | None | TCP, all else normal | Reduced | Normal |
| B | II.2 | F | C140R het* | WGS | Y | 83 | 10.2 | None | TCP, all else normal | Reduced | Normal |
| B | II.3 | F | WT | Sanger | N | 257 | 10.9 | Normal | |||
| C | II.3 | M | G134E het | WGS | N | 253† | 12.5 | ||||
| C | II.4 | F | WT | Sanger | N | 327 | 11.4 | ||||
| C | III.1 | M | G134E het | Sanger | Y | 128 | 9.4 | ||||
| C | III.3 | F | G134E het | WGS | Y | 82 | 8.6 | None | TCP, all else normal | ||
| C | III.4 | M | G134E het | Sanger | N | 184 | 11.1 | ||||
| C | IV.1 | M | G134E het | Sanger | Y | 82 | 8.8 | ||||
| D | I.1 | M | N | ||||||||
| D | I.2 | F | Y | ||||||||
| D | II.2 | F | Y | 110 | 8.2 | ||||||
| D | II.3 | F | Y89C het | Sanger | Y | 97 | 8.9 | Normal | |||
| D | III.1 | M | N | 195 | 9 | ||||||
| D | III.2 | F | Y89C het | WES | Y | 70 | 8.3 | ||||
| E | I.2 | F | Y | ||||||||
| E | II.1 | M | N | 255 | |||||||
| E | II.2 | M | Y | 155 | |||||||
| E | II.3 | M | R96W het | WES | Y | 107 | 9.5 | None | Normal | Reduced | Normal |
| E | II.4 | F | WT | Sanger | N | 220 | 9 | ||||
| E | II.6 | M | R96W het | Sanger | Y | 111 | 10.4 | None | Normal | Reduced | Normal |
| E | II.8 | F | R96W het | Sanger | Y | 104 | 11 | Mild | Normal | Reduced | Normal |
| E | III.1 | F | R96W het | Sanger | Y | 96 | 8.9 | None | |||
| E | III.2 | M | R96W het | Sanger | Y | 110 | 9.4 | None | |||
| E | III.3 | F | Y | 127 | 11 | ||||||
| E | III.4 | M | Y | 135 | 8.8 | ||||||
| E | III.5 | F | Y | 106 | 12 | ||||||
| E | III.6 | M | Y | 140 | 11.9 | ||||||
| E | III.7 | F | R96W het | Sanger | Y | 117 | 11.4 | Mild | Normal | Reduced | Normal |
| E | III.9 | M | N | 275 | |||||||
| E | IV.1 | F | Y | 110 |
Blank entries are not known/not applicable.
BD, bleeding diathesis (none/mild/severe); F, female; het: heterozygous; M, male; MPV, most recent mean platelet volume; N, no; PLT, the mean of PLT measurements is shown where multiple counts had been obtained; TCP, thrombocytopenia; WT: homozygous for the reference allele; Y, yes.
de novo mutation.
PLT measured only after splenectomy to correct thrombocytopenia.
Platelets from IKZF5 cases are depleted of α and dense granules and have an inconsistent aggregation defect
Imaging by light microscopy of blood smears from the probands of pedigrees A, B and E showed no structural abnormalities of platelets and no visible morphological defects in other blood cell types (Figure 2A; Table 1). Transmission EM analysis of platelets from 7 cases and 5 unrelated controls showed that, in keeping with previous measurements of mean platelet volume obtained by hematology analyzers, the platelet size distributions of the cases were normal (Figure 2B-C; Table 1). However, the platelets of the cases were significantly depleted of α granules (Figure 2B,D) and they had empty vacuoles and empty membrane structures (supplemental Figure 1, available on the Blood Web site). Whole mount EM analysis revealed a small but significant reduction in the number of dense granules per platelet in 2 cases from pedigree E (supplemental Figure 2). We performed platelet light transmission aggregometry (LTA) on the probands of pedigrees B, C, D and E. LTA was completely normal in the cases tested in pedigrees B and C but reduced in response to all agonists in the probands of pedigrees D and E (supplemental Table 1). 2 cases in pedigree E described mild bleeding symptoms but there was no significant bleeding history in any other cases (Table 1). Pregnancies had occurred in 5 cases resulting in 11 vaginal deliveries. No one required platelet supplementation during pregnancy or delivery, although 1 case (E II.8) required red cell transfusion for a postpartum hemorrhage in 1 out of 3 deliveries (Table 1; supplemental Table 2).
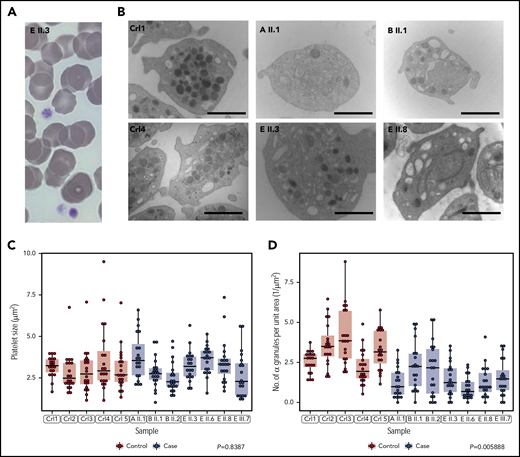
Cases have normally sized platelets but reduced numbers of α granules. (A) Representative May-Grünwald-Giemsa stained peripheral blood smear of patient E II.3. Platelets have a normal appearance (size and granularity) by light microscopy (magnification ×100). (B) Representative EM images for 2 unrelated healthy controls and 4 patients from pedigrees A, B, and E showing depletion of α granules. Marker is 1.5 μM for magnification x20 000. (C, D) The sizes and quantities of α granules per unit area of 20 platelets, quantified blinded to case/control status, in each of 5 cases and 7 controls. Linear mixed model with a fixed effect for case/control status, a random effect for pedigree (groupings in gray) and a random effect for sample. The P value under the null hypothesis that the fixed effect is equal to 0 is shown for each of the 2 responses.
Cases have normally sized platelets but reduced numbers of α granules. (A) Representative May-Grünwald-Giemsa stained peripheral blood smear of patient E II.3. Platelets have a normal appearance (size and granularity) by light microscopy (magnification ×100). (B) Representative EM images for 2 unrelated healthy controls and 4 patients from pedigrees A, B, and E showing depletion of α granules. Marker is 1.5 μM for magnification x20 000. (C, D) The sizes and quantities of α granules per unit area of 20 platelets, quantified blinded to case/control status, in each of 5 cases and 7 controls. Linear mixed model with a fixed effect for case/control status, a random effect for pedigree (groupings in gray) and a random effect for sample. The P value under the null hypothesis that the fixed effect is equal to 0 is shown for each of the 2 responses.
Structural modeling of IKZF5 mutations
Although a crystal structure for IKZF5 (Pegasus) does not exist, the sequence covering Znfs 4 through 7 of the human CCCTC-binding factor CTCF33 is 45% identical to IKZF5 over the 78 residues from C84 to H161 and contains no insertions or deletions. The relevant part of the bound DNA sequence in this structure, AGCAGGGGGC, conforms to the pattern identified for sequences bound by the IKZF5/Pegasus family, nGnnGnnnGn,14 so similar interactions with DNA are expected to be conserved. We found a clear structural explanation for deleterious effects for 3 of the 5 mutations: Y89C would disrupt a binding contact with a backbone phosphate of the DNA; C140R would remove the first ligand in the third Znf domain; and R96W would disrupt a direct interaction with the penultimate guanine base of the consensus sequence in the major groove (Figure 3A). G134 is part of the most common consensus linker sequence between consecutive Znfs (TGEKP35 ) and it may play a role in flexibility during DNA recognition36 and as a recognition site for threonine (or serine) phosphorylation during mitosis,37 so mutations here would be expected to alter function. Although the structure of CTCF reveals no obvious reasons why H155Y could not be accommodated, the surrounding sequence SHHRRR is conserved in almost all of the top 50 hits from our BLAST38 search, but is replaced by KMHILQ in CTCF, which suggests that H155 may be important for interaction with the remainder of IKZF5 or its binding partners. Our modeling thus predicted alterations in Znf binding to DNA for at least 3 of the variants, and we therefore expected alterations in cellular localization of IKZF5 in some of the mutants.

Structural modeling of the variants and expression studies in HEK293 cells. (A) Left panel: Mutation sites in Znf 1 of IKZF5. The structure of Znf 4 from CTCF (PDB entry 5kkq33 ) is portrayed as a template to understand the effect of the mutations Y89C (site equivalent to Y358) and R96W (site equivalent to K365). To model the conformation of R96, K365 in the CTCF structure was replaced by an arginine in a similar conformation to that of R369 in PDB entry 5kl6.34 WT side chains are shown in ball-and-stick representation, whereas the mutated side chains are shown as partially transparent cylinders. Right panel: The role of C140 in Znf 3. We used the coordinates for Znf 6 from PDB entry 5kkq without any changes as all the side-chains shown are conserved between the 2 sequences. (B) Western blot analysis of HEK293 transfected cells with myc-tagged IKZF5 WT and mutants after extraction of chromatin-bound proteins. HIST3H3 (histone H3) serves as loading control. HEK293 transfected with empty vector (Mock) express no IKZF5. The 5 causal mutants are highlighted in red. (C) Quantification of chromatin-bound IKZF5 after transfections and normalization against HIST3H3. The 5 causal mutants are highlighted in red. (D) Immunofluorescence (z-stack images by a structured illumination microscope) using an antibody against IKZF5 (green), phalloidin (actin; red), and 4′,6-diamidino-2-phenylindole (nucleus, blue) showing punctate, nuclear staining for WT IKZF5, whereas R96W and other mutants (supplemental Figure 4) remain largely outside the nucleus after transfecting HEK293 cells. Scale bar, 10 µm.
Structural modeling of the variants and expression studies in HEK293 cells. (A) Left panel: Mutation sites in Znf 1 of IKZF5. The structure of Znf 4 from CTCF (PDB entry 5kkq33 ) is portrayed as a template to understand the effect of the mutations Y89C (site equivalent to Y358) and R96W (site equivalent to K365). To model the conformation of R96, K365 in the CTCF structure was replaced by an arginine in a similar conformation to that of R369 in PDB entry 5kl6.34 WT side chains are shown in ball-and-stick representation, whereas the mutated side chains are shown as partially transparent cylinders. Right panel: The role of C140 in Znf 3. We used the coordinates for Znf 6 from PDB entry 5kkq without any changes as all the side-chains shown are conserved between the 2 sequences. (B) Western blot analysis of HEK293 transfected cells with myc-tagged IKZF5 WT and mutants after extraction of chromatin-bound proteins. HIST3H3 (histone H3) serves as loading control. HEK293 transfected with empty vector (Mock) express no IKZF5. The 5 causal mutants are highlighted in red. (C) Quantification of chromatin-bound IKZF5 after transfections and normalization against HIST3H3. The 5 causal mutants are highlighted in red. (D) Immunofluorescence (z-stack images by a structured illumination microscope) using an antibody against IKZF5 (green), phalloidin (actin; red), and 4′,6-diamidino-2-phenylindole (nucleus, blue) showing punctate, nuclear staining for WT IKZF5, whereas R96W and other mutants (supplemental Figure 4) remain largely outside the nucleus after transfecting HEK293 cells. Scale bar, 10 µm.
Abnormal nuclear localization and chromatin binding in IKZF5 cases
To further investigate the effect of the IKZF5 variants on DNA binding, we expressed the 5 causal mutants, the variant encoding S200G, a low-frequency variant (175 mutant alleles out of 280 830 in gnomAD) encoding I98V in the third Znf domain and WT IKZF5 in HEK293 cells, which do not express this TF. In contrast to WT IKZF5, which enters the nucleus to bind to chromatin, all 5 causal mutants exhibited a strong reduction in chromatin binding (Figure 3B-D), remaining instead in the cytosol and soluble membranes fractions (supplemental Figures 3 and 4). However, S200G and I98V entered the nucleus in a similar manner to WT IKZF5, suggesting that only missense substitutions of highly conserved residues in the N-terminal Znfs may cause thrombocytopenia (Figure 3B-C; supplemental Figure 3).
IKZF5 cases have abnormal PPF
Modeling and cellular localization studies of IKZF5 mutants pointed to defective transcriptional regulation. Because thrombocytopenia can be caused by alterations of TF activity during megakaryopoiesis, we studied MK development in IKZF5 cases. Microscopic evaluation of bone marrow aspirate smears from B II.2, D III.3, E II.8, and E III.2 showed a normal number of MKs without dysmegakaryopoiesis, which suggests normal maturation and differentiation of the megakaryocytic lineage. No abnormalities were present in the other lineages. We further studied MK development using blood-derived HSCs. MK differentiation evaluated by quantifying CD41+ and CD42b+ cells was comparable between subject E II.3 and 2 unrelated controls (Figure 4A). However, PPF by MK in these cultures seemed reduced for E II.3 compared with controls (Figure 4B). To further characterize the PPF defect, we quantified PPF after MK spreading on fibrinogen and staining with VWF and found it to be significantly reduced in 7 cases from 3 pedigrees compared with 6 unrelated healthy controls (P = .0069, linear mixed regression model; Figure 4C). However, there was no discernible difference in the number or spatial distribution of VWF+ α granules in the MKs of cases compared with controls (supplemental Figure 5).
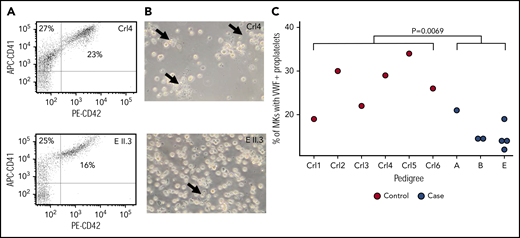
Reduced PPF in MKs from cases. (A) Flow cytometry analysis of day 10 MK for markers CD41 and CD42 showed no difference in maturation between control and case MKs. (B) The same cultures showed evidence of reduced PPF (arrows). (C) PPF was quantified after MK spreading on fibrinogen and staining for VWF for 6 unrelated controls and 7 cases. Linear mixed model with a fixed effect for case/control status, a random effect for pedigree and a random effect for sample. The P value under the null hypothesis in which that the fixed effect is equal to 0 is shown.
Reduced PPF in MKs from cases. (A) Flow cytometry analysis of day 10 MK for markers CD41 and CD42 showed no difference in maturation between control and case MKs. (B) The same cultures showed evidence of reduced PPF (arrows). (C) PPF was quantified after MK spreading on fibrinogen and staining for VWF for 6 unrelated controls and 7 cases. Linear mixed model with a fixed effect for case/control status, a random effect for pedigree and a random effect for sample. The P value under the null hypothesis in which that the fixed effect is equal to 0 is shown.
MK-specific transcriptional regulation if IKZF5
IKZF5 is expressed across hematopoietic lineages but IKZF5 cases have normal blood counts, except for thrombocytopenia. To understand this, we performed RNA-sequencing on the CD4+ T cells, monocytes, neutrophils, and platelets in 3 unrelated cases from pedigrees A, B, and E and 14 unrelated controls. We generated a mean of 51.3M paired-end reads per sample (range, 24.2M-83.9M), of which we aligned a mean of 29.4M (range, 15.4M-53.1M) to the reference human transcriptome (supplemental Figure 6). Our analysis revealed striking differences in the platelet transcriptome landscape only: 1194 protein-coding genes of 10 237 genes in Reactome39 were differentially expressed in platelets but only 4 such genes were differentially expressed in each of the 3 other cell types (Figure 5A). The 631 downregulated, differentially expressed genes (DEGs) in platelets were significantly enriched for pathways involved in platelet function, hemostasis, and membrane and vesicle-mediated transport (Figure 5B), whereas upregulated DEGs had more general functions (supplemental Table 3). The most significantly enriched pathway among the downregulated DEGs was platelet activation, signaling and aggregation, for which 52 of 262 (19.8%) genes were significantly downregulated, including known disease-associated genes2 involved in platelet formation (CDC42, GP1BA, GP9, MPIG6B) and function (FERMT3, PLA2G4A, P2RY12, and TBXA2R) (Figure 5C).
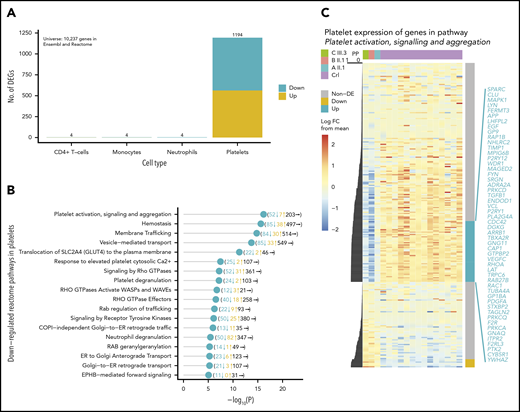
IKZF5 deficiency misregulates transcription in MKs. (A) Barplot of the number of up- and downregulated Reactome/Ensembl coding genes in CD4+ T cells, monocytes, neutrophils, and platelets that are differentially expressed between 14 unrelated controls and 3 unrelated cases. (B) Enriched downregulated pathways in platelets with P < 10−5. The number of downregulated, upregulated, and nondifferentially expressed genes in each pathway are shown in brackets. (D) Heatmap of expression for genes in the “platelet activation, signaling, and aggregation” Reactome pathway. The expression levels have been row-normalized such that the color represents the log FC of expression between samples and the mean log expression weighted by the number of cases and controls. The cases are on the left-most 3 columns and the controls are on the right-most 14 columns. The PP of association is shown to the left of the heatmap and the significantly down- (blue) and upregulated (yellow) genes are shown to the right of the heatmap. The significantly downregulated genes are listed in blue. FC, fold change.
IKZF5 deficiency misregulates transcription in MKs. (A) Barplot of the number of up- and downregulated Reactome/Ensembl coding genes in CD4+ T cells, monocytes, neutrophils, and platelets that are differentially expressed between 14 unrelated controls and 3 unrelated cases. (B) Enriched downregulated pathways in platelets with P < 10−5. The number of downregulated, upregulated, and nondifferentially expressed genes in each pathway are shown in brackets. (D) Heatmap of expression for genes in the “platelet activation, signaling, and aggregation” Reactome pathway. The expression levels have been row-normalized such that the color represents the log FC of expression between samples and the mean log expression weighted by the number of cases and controls. The cases are on the left-most 3 columns and the controls are on the right-most 14 columns. The PP of association is shown to the left of the heatmap and the significantly down- (blue) and upregulated (yellow) genes are shown to the right of the heatmap. The significantly downregulated genes are listed in blue. FC, fold change.
Discussion
Rare variants in other genes cause a combination of thrombocytopenia and α granule abnormalities. Rare variants in NBEAL2 can cause Gray platelet syndrome (GPS), an autosomal recessive disease hallmarked by a complete absence of α granules and an enlargement of platelets, in addition to thrombocytopenia.40,41 In the TF domain, GATA1 mutations cause a paucity of α granules numbers and enlarged platelets,21 GFI1B mutations cause a paucity of α granules and enlarged platelets,42 FLI1 mutations cause fused and enlarged α granules43 and RUNX1 mutations cause α granule storage and secretion defects.44 Interestingly, our RNA-sequencing analysis from platelets lacking functional IKZF5 identified a striking platelet-specific defect, which is consistent with the isolated thrombocytopenia seen in the affected cases. Given the apparent lack of abnormalities in any cell types other than platelets, this may be the first TF-associated thrombocytopenia, which is completely lineage specific.
Transcriptome analysis identified hundreds of DEGs related to membrane and vesicle-mediated transport pathways, pointing to a possible mechanism for the α granule defect. IKZF5-related disorder does not appear to lead to a significant platelet function defect, as shown by the lack of bleeding symptoms in the majority of cases. Platelet function results obtained by light transmission aggregometry were inconsistent among the cases, with some having normal responses to all agonists. This variability is similar to that seen in other α granule defects such as GPS45 ; however, further studies of platelet function are required to understand the clinical implication, if any, of the observed paucity of α granules. In contrast to GPS, there was no clinical evidence of myelofibrosis (such as splenomegaly, other cytopenias, or morphological changes on the blood smear) in any of the cases presented here and, unlike the other Ikaros proteins, there is no evidence of lymphoid or immune dysregulation. Therefore, based on the available evidence, thrombocytopenia resulting from rare missense variants in IKZF5, is associated with a good prognosis, in sharp contrast with thrombocytopenia from mutations in other TFs such as RUNX1 and ETV6. The reduction in PLT in IKZF5 cases is mild: none of the PLTs in members of the 5 pedigrees were <70 × 109/L and 1 unrecallable carrier had a PLT as high as 184 × 109/L, reflecting the complex regulation of megakaryopoiesis and the ensuing variation in PLT in the normal population. However, there was no evidence of alterations in mean platelet volume in IKZF5 cases compared with their healthy relatives.
Heterozygous mutations in the DNA-binding Znf domains of IKZF1 exert a dominant-negative effect,46,47 with interactions between the WT and mutated DNA-binding domains resulting in dimers that lack transcriptional activity.48 We propose that the mechanism through which the heterozygous variants in IKZF5 exert their effect is similar. We have shown that the IKZF5 variants cause abnormal cellular localization and decreased chromatin binding in HEK293T cells and, in vivo, this would be expected to affect transcription. The discovery of rare missense variants in the N-terminal Znf domains of IKZF5 as a novel cause of inherited thrombocytopenia highlights an important role for this previously unknown TF in megakaryocyte and platelet biology.
The RNA-seq data reported in this article have been deposited at the European Genome phenome Archive (accession number ID EGAD00001005107).
The online version of this article contains a data supplement.
The publication costs of this article were defrayed in part by page charge payment. Therefore, and solely to indicate this fact, this article is hereby marked “advertisement” in accordance with 18 USC section 1734.
Acknowledgments
The authors thank Ma Hazel Cordial for assisting in patient sampling and Katherine Wedderburn and Lindsay Walker for international sample handling; NIHR BioResource volunteers and their families for their participation, and gratefully acknowledge NIHR BioResource centers and National Health Service (NHS) Trusts and staff for their contribution; and NIHR and NHS Blood and Transplant.
Funding for the project was provided by the NIHR (RG65966). C.L. and M.C.S. received funding from MRC Clinical Research Training Fellowships (MR/J011711/1 and MR/R002363/1). K.F. is supported by the Research Council of the University of Leuven (BOF KU Leuven, Belgium, OT/14/098) and grants from CSL Behring, Bayer, and SOBI. M.F. is supported by the British Heart Foundation (FS/18/53/33863). D.S.’s work has been supported in part by an Isaac Newton fellowship to M.F. S.S. is the recipient of an NIHR Academic Clinical Fellowship.
The views expressed are those of the author(s) and not necessarily those of the NHS, the NIHR, or the Department of Health.
Authorship
Contribution: E.T., K.F., C.L., and S.S. wrote the manuscript; D.G., D.S., R.J.R., and K.M. edited the manuscript; C.L., S.S., R.F., S.M., K.S., M.J.S., J.-C.B., C.A.-A., K.G., M.A.L., M.S.C. and K.M. enrolled patients and collected clinical data; S.S., C.L., and K.M. coordinated cosegregation studies; S.P. supported project coordination; C.T., M.S., F.B., L.S., J.C.S., M.C.S., S.F., and K.F. performed experiments; W.E. analyzed blood smears; C.J.P. supervised the whole-genome sequencing (WGS) bioinformatics pipeline; K.E.S. managed WGS logistics; D.G., D.S., N.G., L.G., and E.T. performed data analyses; R.J.R. performed structural modelling; and W.H.O., M.L., M.F., K.F., and E.T. supervised the project.
Conflict-of-interest disclosure: The authors have no conflicts of interest to declare.
A complete list of the members of the NIHR BioResource appears in the supplemental appendix.
Correspondence: Ernest Turro, University of Cambridge, Department of Haematology, Long Rd, Cambridge CB2 0PT, United Kingdom; e-mail: et341@cam.ac.uk.
