

Key Points
DCs are the principal source of IL-6 dysregulation after alloSCT.
IL-6–dependent GVHD is driven by classical signaling of IL-6R on donor T cells but is regulated by trans signaling.

Abstract
Graft-versus-host disease (GVHD) after allogeneic stem cell transplantation (alloSCT) is characterized by interleukin-6 (IL-6) dysregulation. IL-6 can mediate effects via various pathways, including classical, trans, and cluster signaling. Given the recent availability of agents that differentially inhibit these discrete signaling cascades, understanding the source and signaling and cellular targets of this cytokine is paramount to inform the design of clinical studies. Here we demonstrate that IL-6 secretion from recipient dendritic cells (DCs) initiates the systemic dysregulation of this cytokine. Inhibition of DC-driven classical signaling after targeted IL-6 receptor (IL-6R) deletion in T cells eliminated pathogenic donor Th17/Th22 cell differentiation and resulted in long-term survival. After engraftment, donor DCs assume the same role, maintaining classical IL-6 signaling–dependent GVHD responses. Surprisingly, cluster signaling was not active after transplantation, whereas inhibition of trans signaling with soluble gp130Fc promoted severe, chronic cutaneous GVHD. The latter was a result of exaggerated polyfunctional Th22-cell expansion that was reversed by IL-22 deletion or IL-6R inhibition. Importantly, inhibition of IL-6 classical signaling did not impair the graft-versus-leukemia effect. Together, these data highlight IL-6 classical signaling and downstream Th17/Th22 differentiation as important therapeutic targets after alloSCT.
Introduction
Allogeneic stem cell transplantation (alloSCT) is a potent curative therapy for many hematological malignancies that is largely mediated by donor T cell–dependent graft-versus-leukemia (GVL) responses. However, donor alloreactive T cells may also damage nonmalignant host tissue, culminating in graft-versus-host disease (GVHD). GVHD can develop in both acute and chronic forms, manifesting within the gastrointestinal (GI) tract, skin, and liver, resulting in significant morbidity and mortality.1
GVHD is initiated by conditioning-induced tissue damage, with subsequent compromise of the mucosal barrier in the GI tract, facilitating translocation of microbiome-derived pathogen-associated molecular patterns driving systemic interleukin-6 (IL-6) dysregulation.2-4 IL-6 is a pleiotropic cytokine that can be secreted by most cell types and is responsible for orchestrating expansion and differentiation of multiple myeloid and T-cell lineages.5,6 A clear pathogenic role has been established for IL-6 in many inflammatory disorders, and its inhibition is associated with the attenuation of symptoms in patients with rheumatoid arthritis, juvenile idiopathic arthritis, and Castleman’s disease and those undergoing alloSCT.7-13 However, IL-6 is also an important driver of homeostatic processes, such as tissue repair, and its inhibition is associated with an increased incidence of intestinal perforations, dyslipidemia, elevated liver transaminases, and transient neutropenia in patients.14,15 These limitations have led to the development of several classes of IL-6 inhibitors that can differentially inhibit the complex signaling cascades of IL-6.16
IL-6 can signal via at least 3 pathways, classical, trans, and the recently described cluster signaling, with each pathway forming a distinct complex of IL-6, the ligand-specific IL-6 receptor α (IL-6Rα), and the common gp130 signal transducer on target cells.5,17,18 IL-6 classical signaling involves binding of IL-6 to the membrane-bound IL-6R (mIL-6R), leading to recruitment and homodimerization of gp130. Alternatively, IL-6 trans signaling utilizes a soluble form of IL-6R (sIL-6R) and therefore only requires surface expression of gp130 by target cells. Similarly, IL-6 cluster signaling involves internal formation of an IL-6/IL-6R complex by dendritic cells (DCs) that is transpresented to gp130 on T cells within the immunological synapse in the context of antigen presentation.17,18
In the hematology setting, IL-6 and subsequent phosphorylation of STAT3 have emerged as major targets in the treatment of complications of immunotherapy, for both GVHD after alloSCT and cytokine release syndrome after chimeric antigen receptor T-cell infusion. Therefore, direct IL-6 inhibition shows significant activity in preventing and/or treating both complications.11,12,19,20 Similarly, inhibitors of STAT phosphorylation are increasingly used in transplantation, with ruxolitinib now US Food and Drug Administration approved for the treatment of steroid-refractory acute GVHD (aGVHD). With this in mind, it is imperative to understand the pathogenic signaling cascades used by IL-6 to logically test new targeted therapeutics. In preclinical murine models of alloSCT, prophylactic inhibition of all 3 IL-6 pathways with anti–IL-6R antibody has shown promise in attenuating aGVHD.21,22 Moreover, this effect has largely been attributed to the critical role of IL-6 in driving pathogenic T-cell populations.21,23-31 This reduction in aGVHD was also reflected in 2 phase 2 clinical trials of the anti–IL-6R antibody tocilizumab for the prophylaxis of aGVHD, and a phase 3 study has also been undertaken (ACTRN12614000212651).11,12 The relevant signaling pathways by which IL-6 mediates alloimmunity have not been characterized, and given that multiple clinical IL-6 inhibitors are available, this is critical information for the design of clinical studies. Here we show that recipient DCs are the primary source of IL-6 dysregulation and that the classical pathway is responsible for GVHD and mortality.
Methods
Mice
All mice used in this study were female, and the strains used are listed in supplemental Table 1, available on the Blood Web site. Mice were housed in sterilized microisolator cages and received acidified autoclaved water (pH, 2.5) alone, autoclaved water containing enrofloxacin (Baytril; Provet; 100 mg/L; B6 wild-type and soluble gp130Fc (sgp130Fc) recipients; first 2 weeks posttransplantation), or neomycin (1 g/L; CD11cCre and VillinERT crosses as recipients; 2 weeks before transplantation and maintained thereafter). For cohousing experiments, mice were cohoused in large cages for 4 weeks before transplantation and maintained together for the duration of the experiment. All animal experiments were approved by and performed in accordance with the QIMR Berghofer Animal Ethics Committee.
Stem cell/BM transplantation
Recipient mice underwent either 900- (BALB/c), 1000- (B6), or 1100-cGy (B6D2F1) total-body irradiation (caesium-137 source), unless otherwise stated, split over 2 doses (day 1) 3 hours apart. Mice received grafts containing bone marrow (BM) alone (non-GVHD) or with T cells enriched by negative BioMag (Qiagen) bead depletion of nonTcells or fluorescence-activated cell sorting (CD90.2+7AAD−). Non-GVHD control groups received T cell–depleted (TCD) BM grafts. Mice were monitored daily and systemic GVHD assessed weekly using a cumulative scoring system based on weight loss, posture, mobility, fur texture, and skin integrity (maximum score of 2 per parameter to give a maximum total of 10).32 Mice with GVHD scores ≥6 were euthanized and date of death registered as the following day. For histology, tissue samples were paraformaldehyde fixed and paraffin embedded before sections were cut and stained with hematoxylin and eosin.
To induce Cre recombinase expression in Villin-CreERT2 mice, Villin-Cre+ × IL-6Rfl/fl (IL-6Rfl) and Villin-Cre− × IL-6Rfl control mice were cohoused and injected with tamoxifen (MP Biomedicals) for 5 consecutive days (1 mg per day intraperitoneally) and underwent transplantation 2 weeks after the first injection.33,34
For GVL studies, BCR-ABL/NUP98-HOXA9 (B6D2F1, GFP+, H-2Dd/b, CD45.2+)35 and MLL-AF9 (B6D2F1, GFP+, H-2Dd/b, CD45.2+)36 leukemias were used. Irradiated recipient mice received grafts containing leukemia and B6 TCD BM alone or with BioMag-purified T cells. Mice with a high leukemia burden (white blood cell count >50 × 106 or >30% leukocytes GFP+), paralysis, or clinical scores ≥6 were euthanized.
Bioluminescence imaging
Mice were subcutaneously injected with 500 μg of D-luciferin (PerkinElmer), and 5 minutes after injection, luciferase signal intensity was analyzed for T-cell expansion (Xenogen IVIS 100; Caliper Life Sciences).
Antibodies
The antibodies used in this study are provided in supplemental Table 2. For IL-6R blockade studies, rat anti-mouse IL-6R monoclonal antibody (MR16-1; Chugai Pharmaceutical Co.) was administered intraperitoneally at 500 μg per dose on day 1 and days +3 and +7 posttransplantation as described.22 Rat immunoglobulin G was used as an isotype control (Sigma-Aldrich).
Serum soluble receptor and cytokines
Soluble receptor levels were assessed by commercial enzyme-linked immunosorbent assay kits (R&D Systems): murine sIL-6R (Mouse IL-6R α DuoSet ELISA), sgp130 (Mouse gp130 DuoSet ELISA), and sgp130Fc (Human sgp130 DuoSet ELISA; with recombinant human sgp130Fc chimera protein [R&D Systems] as the standard). Serum cytokine levels were measured by Flex Array (BD Biosciences).
Cell preparation and intracellular analysis
For all intracellular cytokine staining, cells were stimulated for 4 hours with phorbol 12-myristate 13-acetate (5 μg/mL) and ionomycin (50 μg/mL; Sigma-Aldrich) in the presence of brefeldin A (BioLegend) at 37°C with 5% carbon dioxide at >95% humidity. Cells were surface labeled, fixed, and permeabilized using the Cytofix/Cytoperm kit (BD Biosciences) for cytokines and the Fixation and Permeabilization Buffer set (eBioscience) for transcription factors. Quantification of STAT3 phosphorylation was performed as previously described.37 All samples were acquired via LSR Fortessa (BD Biosciences) and analyzed via FlowJo v9 software.
Gene expression analysis
Naïve B6 wild-type mice were irradiated (1000 cGy) and cell populations sort purified from the mesenteric lymph nodes (mLNs) 8 hours later. Total RNA was extracted using the RNeasy Micro Kit (Qiagen), and complementary DNA was prepared with the Maxima H Minus First Strand cDNA Synthesis Kit (Thermo Fisher). Gene expression was determined by quantitative polymerase chain reaction using TaqMan GE assays (Applied Biosystems) for murine IL6 (Mm00446190_m1), in parallel with the housekeeping gene Hprt (Mm03024075_m1). IL6 expression was determined using the comparative Ct method (2−ΔΔCt) normalized relative to Hprt.
Statistical analysis
Log-rank (Mantel-Cox) tests were used for all nonparametric murine survival data, with a Mann-Whitney test for clinical scores at specified time points. A 1-way analysis of variance Holm-Sidak’s multiple comparison test to a single pooled variance was used when comparing data with ≥3 groups for serum cytokine levels, T-cell differentiation, and serum cytokine time course experiments. Unpaired Student t tests were used for serum protein levels and serum cytokine and T-cell cytokine expression with normally distributed data. Unpaired 2-tailed Mann-Whitney tests were used to evaluate differences in histopathology analyses and nonparametric T-cell cytokine expression. Data are presented as mean ± standard error of the mean, and P < .05 was considered statistically significant. Statistical analyses were performed using GraphPad Prism v7.02.
Results
Systemic IL-6 dysregulation originates in recipient DCs
IL-6 is systemically dysregulated after allogeneic BM transplantation (alloBMT),11,21,22 but because of the almost ubiquitous expression of IL-6, the important cellular sources remain unknown. We investigated the role of recipient DCs in this process (Figure 1A-B), because IL-6 is primarily derived from host cells after SCT, and DCs are important in activating T cells after BMT.22,38 Surprisingly, despite the broad cellular expression of IL-6, systemic IL-6 dysregulation was critically dependent on recipient DCs alone (Figure 1C). IL-6 dysregulation was dependent on both conditioning intensity and donor T-cell dose (supplemental Figure 1), with a major contribution from recipient CD11b+CD103+ DCs that exhibited high IL-6 messenger RNA levels after irradiation (Figure 1D-E). To confirm this, we quantified the differentiation of IL-6–dependent donor T-cell subsets in the mLNs and spleen of recipients in which IL-6 or IL-6R had been deleted in DCs only or IL-6 deleted in all recipient tissue. Consistent with the systemic IL-6 levels in these recipients, recipient DC-derived IL-6 was critical for directing differentiation of donor CD4+ T cells into pathogenic Th17 (CD4+IL-17+T) and Th22 (CD4+IL-17−IL-22+T) subsets, and defects were comparable to those seen in IL-6−/− recipients (Figure 1F-H). This effect was specific to IL-6–dependent subsets, because IL-6 was not required for IFNγ, TNF, GM-CSF, or IL-10 cytokine expression in CD4+ T cells (Figure 1I-J), and it could not be attributed to altered APC development, because myeloid development was normal in the Cre-lox recipient combinations used (supplemental Figure 2). The expression of IL-6R by recipient DCs was unnecessary for donor Th17 and Th22 differentiation, confirming that IL-6 signaling to DCs and the transpresentation of IL-6/IL-6R complexes by DCs (ie, cluster signaling) were not required to induce donor T-cell differentiation.
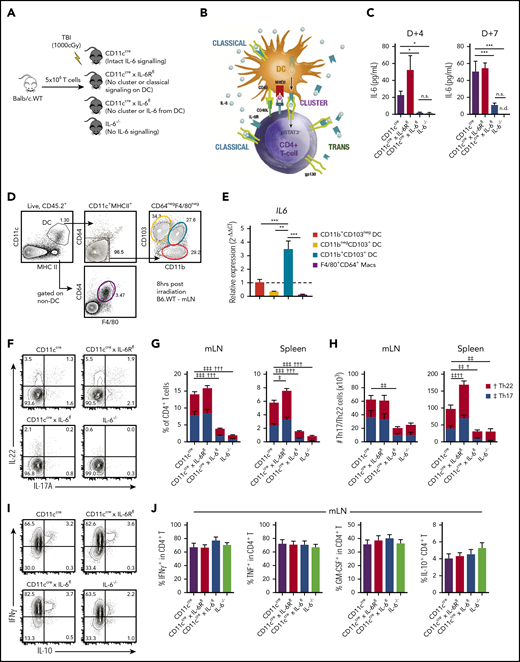
Systemic IL-6 dysregulation originates in recipient DCs. (A,C,F-J) Lethally irradiated recipient CD11cCre (intact IL-6 signaling), CD11cCre × IL-6Rfl (DCs are deficient in classical and unable to induce cluster signaling), CD11cCre × IL-6fl (DCs are deficient in IL-6 and unable to induce cluster signaling), and IL-6−/− (all recipient cells are deficient in IL-6) mice received transplants of 5 × 106 T cells from wild-type BALB/c (BALB/c.WT) donors. (B) DCs can signal via IL-6 or induce IL-6 signaling to gp130 on T cells through 3 pathways: classical (involving DC-derived IL-6), trans (requiring DC-derived IL-6 ligated to any source of sIL-6R), and cluster (requiring DC-derived IL-6 and IL-6R). (C) Peripheral blood serum levels of IL-6 from recipients at day 4 and day 7 after transplantation (n = 5-8 mice per group from 2 experiments). (D-E) B6.WT mice were irradiated (1000 cGy), and 8 hours later, CD103+CD11b−, CD103+CD11b+, and CD103−CD11b+ DCs (CD11c+MHCIIhiCD64−F4/80−) and non-DC boolean-gated CD64+F4/80+ macrophages were sort purified from the mLNs (D) and IL6 mRNA levels measured by quantitative polymerase chain reaction and quantified relative to CD103+CD11b− DCs (n = 3, each pooled from mLNs from 2-5 mice) (E). (F) Representative flow cytometry plots of IL-22 and IL-17A expression in CD4+ T cells from the mLNs (concatenated from 4 mice per group) and stacked bar graphs of the frequency (G) and total numbers (H) of donor Th17 (CD4+IL-17A+) and Th22 (CD4+IL-22+IL-17A−) cells in the mLNs and spleen at day 7 posttransplantation (n = 4-14 mice per group from 2-3 experiments). (I-J) Representative flow cytometry plots (I) and frequency (J) of interferon-γ (IFNγ), tumor necrosis factor (TNF), granulocyte-macrophage colony-stimulating factor (GM-CSF), and IL-10 expression in CD4+ T cells from the mLNs (concatenated from 4 mice per group; n = 8-14 mice per group from 3 experiments). Data presented as mean ± standard error of the mean. *P < .05, **P < .01, ***P < .001. †P < .05, ††P < .01, †††P < .001 for Th22. ‡P < .05, ‡‡P < .01, ‡‡‡P < .001 for Th17. MHC, major histocompatibility complex; n.d., not detectable; n.s., not significant; TBI, total-body irradiation.
Systemic IL-6 dysregulation originates in recipient DCs. (A,C,F-J) Lethally irradiated recipient CD11cCre (intact IL-6 signaling), CD11cCre × IL-6Rfl (DCs are deficient in classical and unable to induce cluster signaling), CD11cCre × IL-6fl (DCs are deficient in IL-6 and unable to induce cluster signaling), and IL-6−/− (all recipient cells are deficient in IL-6) mice received transplants of 5 × 106 T cells from wild-type BALB/c (BALB/c.WT) donors. (B) DCs can signal via IL-6 or induce IL-6 signaling to gp130 on T cells through 3 pathways: classical (involving DC-derived IL-6), trans (requiring DC-derived IL-6 ligated to any source of sIL-6R), and cluster (requiring DC-derived IL-6 and IL-6R). (C) Peripheral blood serum levels of IL-6 from recipients at day 4 and day 7 after transplantation (n = 5-8 mice per group from 2 experiments). (D-E) B6.WT mice were irradiated (1000 cGy), and 8 hours later, CD103+CD11b−, CD103+CD11b+, and CD103−CD11b+ DCs (CD11c+MHCIIhiCD64−F4/80−) and non-DC boolean-gated CD64+F4/80+ macrophages were sort purified from the mLNs (D) and IL6 mRNA levels measured by quantitative polymerase chain reaction and quantified relative to CD103+CD11b− DCs (n = 3, each pooled from mLNs from 2-5 mice) (E). (F) Representative flow cytometry plots of IL-22 and IL-17A expression in CD4+ T cells from the mLNs (concatenated from 4 mice per group) and stacked bar graphs of the frequency (G) and total numbers (H) of donor Th17 (CD4+IL-17A+) and Th22 (CD4+IL-22+IL-17A−) cells in the mLNs and spleen at day 7 posttransplantation (n = 4-14 mice per group from 2-3 experiments). (I-J) Representative flow cytometry plots (I) and frequency (J) of interferon-γ (IFNγ), tumor necrosis factor (TNF), granulocyte-macrophage colony-stimulating factor (GM-CSF), and IL-10 expression in CD4+ T cells from the mLNs (concatenated from 4 mice per group; n = 8-14 mice per group from 3 experiments). Data presented as mean ± standard error of the mean. *P < .05, **P < .01, ***P < .001. †P < .05, ††P < .01, †††P < .001 for Th22. ‡P < .05, ‡‡P < .01, ‡‡‡P < .001 for Th17. MHC, major histocompatibility complex; n.d., not detectable; n.s., not significant; TBI, total-body irradiation.
Donor DC–derived IL-6 maintains alloreactive T-cell expansion and differentiation
Donor DCs are critical for alloantigen presentation and cytokine secretion within the GI tract after transplantation.39 Considering the magnitude of the effect invoked by recipient DCs, we investigated the role of donor DCs in promoting IL-6–dependent aGVHD. We used a model whereby recipient BALB/c mice were transplanted with donor B6 grafts in which either IL-6 or IL-6R were specifically deleted (or intact) in DCs. In contrast to recipient DCs, the absence of IL-6 expression by donor DCs did not influence systemic IL-6 levels after transplantation (Figure 2A). In subsequent experiments, donor luciferase–expressing T cells that expressed a transgenic T-cell receptor (TEaluc+) specific for a host I-E–derived peptide (Eα) presented within donor I-Ab were transferred 12 days after BMT, and luciferase was used as a reporter for alloantigen-specific T-cell expansion 3 days later (Figure 2B).39 As we have previously reported, significant and localized allospecific T-cell expansion was noted within the mLNs (Figure 2C-D). This expansion required IL-6 expression by donor DCs but was independent of their IL-6R expression (Figure 2C-D). Confirming this effect, we also observed a significant reduction in TEa T-cell numbers in the mLNs of animals lacking DC-derived IL-6 (Figure 2E), together with reductions in donor Th17, Th22, and Th1 cell numbers (Figure 2F-G).
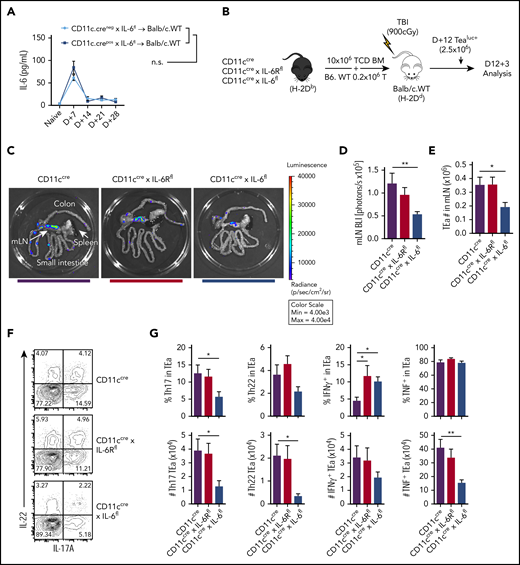
Donor DC–derived IL-6 maintains alloreactive T-cell expansion and differentiation. (A) BALB/c mice were lethally irradiated (900 cGy) and received donor grafts containing 10 × 106 TCD BM and 0.2 × 106 T cells from either CD11c.Cre− × IL-6fl or CD11c.Cre+ × IL-6fl mice, and peripheral blood serum IL-6 levels were monitored over 28 days (n = 8-12 mice per group from 2 experiments). (B) BALB/c mice were lethally irradiated and received transplants of 10 × 106 TCD BM from either CD11cCre, CD11cCre × IL-6Rfl, or CD11cCre × IL-6fl mice, along with 0.2 × 106 B6 wild-type (B6.WT) T cells. On day 12 after transplantation, 2.5 × 106 TEaluc+ cells were injected and 3 days later quantified in the GI tract. (C) Representative images of bioluminescence imaging (BLI) in the GI tract, mLNs, and spleen are shown. (D) Quantification of BLI in the mLNs (n = 13-15 mice per group from 3 experiments). (E) Quantification of total TEaluc+ (CD45.1+Vβ6+Vα2+) cells in the mLNs by flow cytometry. (F-G) Representative contour plots (concatenated from 4 mice per group) (F) and frequency and enumeration (G) of Th17 (IL-17+), Th22 (IL-22+IL-17−), and IFNγ and TNF expression by TEaluc+ cells from the mLNs (n = 9 mice per group from 2 experiments). Data presented as mean ± standard error of the mean. *P < .05, **P < .01. TBI, total-body irradiation.
Donor DC–derived IL-6 maintains alloreactive T-cell expansion and differentiation. (A) BALB/c mice were lethally irradiated (900 cGy) and received donor grafts containing 10 × 106 TCD BM and 0.2 × 106 T cells from either CD11c.Cre− × IL-6fl or CD11c.Cre+ × IL-6fl mice, and peripheral blood serum IL-6 levels were monitored over 28 days (n = 8-12 mice per group from 2 experiments). (B) BALB/c mice were lethally irradiated and received transplants of 10 × 106 TCD BM from either CD11cCre, CD11cCre × IL-6Rfl, or CD11cCre × IL-6fl mice, along with 0.2 × 106 B6 wild-type (B6.WT) T cells. On day 12 after transplantation, 2.5 × 106 TEaluc+ cells were injected and 3 days later quantified in the GI tract. (C) Representative images of bioluminescence imaging (BLI) in the GI tract, mLNs, and spleen are shown. (D) Quantification of BLI in the mLNs (n = 13-15 mice per group from 3 experiments). (E) Quantification of total TEaluc+ (CD45.1+Vβ6+Vα2+) cells in the mLNs by flow cytometry. (F-G) Representative contour plots (concatenated from 4 mice per group) (F) and frequency and enumeration (G) of Th17 (IL-17+), Th22 (IL-22+IL-17−), and IFNγ and TNF expression by TEaluc+ cells from the mLNs (n = 9 mice per group from 2 experiments). Data presented as mean ± standard error of the mean. *P < .05, **P < .01. TBI, total-body irradiation.
IL-6 trans signaling increases late after transplantation and regulates cutaneous GVHD
Having established that host DCs are the major source of systemic IL-6 after transplantation, we next explored the relative contribution of the IL-6 classical and trans signaling pathways to GVHD. The relative balance between IL-6 classical and trans signaling can be inferred by the ratio of systemic sIL-6R (required for trans signaling) and sgp130 (antagonist of trans signaling) levels in conjunction with the presence of IL-6 itself. Thus, higher molar ratios of sIL-6R/sgp130 favor trans signaling, whereas lower sIL-6R levels favor classical signaling.40-42 Levels of IL-6 in sera were not detectable in naïve mice but were increased very early after myeloablative irradiation (Figure 3A-B). In contrast, the serum levels of sIL-6R transiently dropped at day 4 before rising above baseline levels from day 14 (Figure 3C). Levels of sgp130 decreased after transplantation (Figure 3C), such that the relative molar ratio of sIL-6R (∼40 kDa) to sgp130 (∼100 kDa) increased from day 14 after transplantation (Figure 3D). Together, these data suggest that IL-6 classical signaling is favored early after transplantation, whereas trans signaling increases in the later phases.
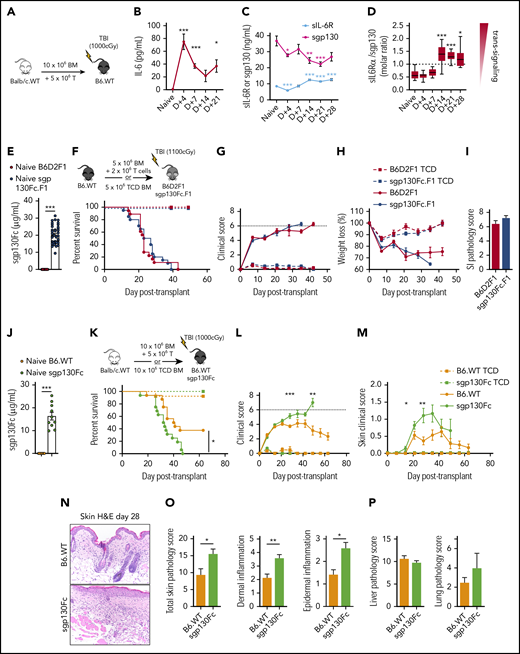
IL-6 trans signaling increases late after transplantation and regulates cutaneous GVHD. (A-D) Lethally irradiated B6 wild-type (B6.WT) mice received 10 × 106 BM and 5 × 106 T-cell grafts from BALB/c mice. Peripheral blood serum levels of IL-6 (A), sIL-6R (required for IL-6 trans signaling) (B), and sgp130 (antagonist of IL-6 trans signaling) (C) were analyzed from naïve B6 mice and B6 mice receiving transplants at the indicated times after transplantation (n = 6-28 mice per group from 1-4 experiments). (D) Molar ratio of sIL-6R (∼40 kDa) to sgp130 (∼100 kDa) from matched samples in panels B and C, with a higher ratio indicating a greater incidence of IL-6 trans signaling (box and Tukey whiskers). (E-O) The role of IL-6 trans signaling in GVHD was assessed in preclinical models of acute (E-I) (B6 → B6D2F1) and chronic (J-O) (BALB/c → B6) GVHD. Transgenic sgp130Fc.F1 (B6D2F1) and sgp130Fc (B6) recipients (E, J) expressed high serum levels of the IL-6 trans signaling inhibitor sgp130Fc before transplantation (n = 10-40 mice per group from 2-4 experiments).43,45 (F-H, K-M) Lethally irradiated mice received BM and T-cell grafts or TCD BM grafts as non-GVHD controls, as shown in the schema (F, K). Survival indices by Kaplan-Meier analyses (F), combined clinical scores (G), and weight loss (H) of B6D2F1 and sgp130Fc.F1 recipients after transplantation (BM and T-cell grafts, n = 18 mice per group; TCD, n = 6 mice per group; from 3 experiments). (I) Quantitative GVHD histopathological analysis of the ileum on day 7 after transplantation (n = 7 mice per group from 2 separate experiments). Survival indices by Kaplan-Meier analyses (K), combined clinical scores (L), and clinical scores (M) of skin pathology after transplantation (BM and T-cell grafts, n = 18 mice per group; TCD, n = 8-11 mice per group; from 3 experiments). (N-P) Lethally irradiated B6 and sgp130Fc mice received 10 × 106 BM and 3 × 106 T-cell grafts from BALB/c.45.1 mice, and tissue was collected 28 days (N-O) or 21 days (P) after transplantation. (N) Representative images of skin histology (200× magnification) and semiquantitative GVHD pathology analysis (O-P) of skin (O) and liver and lung (P) sections (n = 5-7 mice per group from 1 representative experiment). Data presented as mean ± standard error of the mean. *P < .05, **P < .01, ***P < .001. H&E, hematoxylin and eosin; SI, small intestine.
IL-6 trans signaling increases late after transplantation and regulates cutaneous GVHD. (A-D) Lethally irradiated B6 wild-type (B6.WT) mice received 10 × 106 BM and 5 × 106 T-cell grafts from BALB/c mice. Peripheral blood serum levels of IL-6 (A), sIL-6R (required for IL-6 trans signaling) (B), and sgp130 (antagonist of IL-6 trans signaling) (C) were analyzed from naïve B6 mice and B6 mice receiving transplants at the indicated times after transplantation (n = 6-28 mice per group from 1-4 experiments). (D) Molar ratio of sIL-6R (∼40 kDa) to sgp130 (∼100 kDa) from matched samples in panels B and C, with a higher ratio indicating a greater incidence of IL-6 trans signaling (box and Tukey whiskers). (E-O) The role of IL-6 trans signaling in GVHD was assessed in preclinical models of acute (E-I) (B6 → B6D2F1) and chronic (J-O) (BALB/c → B6) GVHD. Transgenic sgp130Fc.F1 (B6D2F1) and sgp130Fc (B6) recipients (E, J) expressed high serum levels of the IL-6 trans signaling inhibitor sgp130Fc before transplantation (n = 10-40 mice per group from 2-4 experiments).43,45 (F-H, K-M) Lethally irradiated mice received BM and T-cell grafts or TCD BM grafts as non-GVHD controls, as shown in the schema (F, K). Survival indices by Kaplan-Meier analyses (F), combined clinical scores (G), and weight loss (H) of B6D2F1 and sgp130Fc.F1 recipients after transplantation (BM and T-cell grafts, n = 18 mice per group; TCD, n = 6 mice per group; from 3 experiments). (I) Quantitative GVHD histopathological analysis of the ileum on day 7 after transplantation (n = 7 mice per group from 2 separate experiments). Survival indices by Kaplan-Meier analyses (K), combined clinical scores (L), and clinical scores (M) of skin pathology after transplantation (BM and T-cell grafts, n = 18 mice per group; TCD, n = 8-11 mice per group; from 3 experiments). (N-P) Lethally irradiated B6 and sgp130Fc mice received 10 × 106 BM and 3 × 106 T-cell grafts from BALB/c.45.1 mice, and tissue was collected 28 days (N-O) or 21 days (P) after transplantation. (N) Representative images of skin histology (200× magnification) and semiquantitative GVHD pathology analysis (O-P) of skin (O) and liver and lung (P) sections (n = 5-7 mice per group from 1 representative experiment). Data presented as mean ± standard error of the mean. *P < .05, **P < .01, ***P < .001. H&E, hematoxylin and eosin; SI, small intestine.
IL-6 trans signaling is reported to drive pathology in other IL-6–dependent inflammatory disorders,5 and a specific inhibitor, sgp130Fc, has been developed and is entering clinical trials.40,43,44 sgp130Fc is a dimeric sgp130 complex fused to an immunoglobulin G1 antibody Fc region that, similar to the naturally occurring sgp130, inhibits IL-6 trans signaling by sequestering IL-6/sIL-6R complexes.43-45 We therefore investigated the impact of IL-6 trans signaling on GVHD by utilizing transgenic mice that expressed high levels of sgp130Fc in preclinical models of aGVHD (where mice develop early GI GVHD; Figure 3E-F) and models in which mice developed features of both aGVHD and chronic GVHD (cGVHD; characterized by fibrosis and skin lesion development26,46 ; Figure 3J-K).
In aGVHD systems, inhibition of IL-6 trans signaling did not affect GVHD mortality, weight loss, or histopathology in the GI tract (Figure 3F-I). In contrast, inhibition of IL-6 trans signaling in a cGVHD model significantly accelerated GVHD mortality and promoted the development of severe cutaneous GVHD (Figure 3K-M). Moreover, the enhanced cutaneous GVHD was reflected by histopathological analysis of skin, but not liver or lung, tissue posttransplantation, with skin tissue exhibiting increased cellular infiltration and inflammation (Figure 3N-P). This effect was specific to GVHD, because non-GVHD recipients receiving TCD grafts did not develop clinical skin GVHD. Collectively, these data indicate that IL-6 trans signaling is active late after BMT and regulates cGVHD.
IL-6 trans signaling regulates Th17 and Th22 differentiation
To understand how IL-6 trans signaling regulates cGVHD, we investigated donor CD4+ T-cell differentiation in the mLNs after transplantation by high-dimensional cytokine analysis. Here we observed that inhibition of IL-6 trans signaling promoted the expression of multiple proinflammatory cytokines (IFNγ, GM-CSF, and IL-22), which contrasted with a striking decrease in IL-10 expression (Figure 4A). Proportionally, the largest increase was in IL-22–expressing CD4+ T cells, which were recently shown to be a potent mediator of skin GVHD.29 Delineation of the source of elevated IL-22 highlighted a marked expansion in Th22 cells in multiple organs, which was most prominent in the mLNs and peripheral LNs of sgp130Fc recipients (Figure 4B-C). In contrast, IL-22+Th17 cells were minimally elevated, with no differences in the IL-22−Th17 population. Moreover, a majority of Th22 cells also coexpressed IFNγ, TNF, and GM-CSF and lacked IL-10 expression, consistent with a polyfunctional and proinflammatory T-cell profile (Figure 4A).29,31 Nevertheless, the increase in IFNγ and GM-CSF expression in CD4+ T cells was primarily within the Th1 population (Figure 4D-E). Furthermore, the reduced expression of IL-10 in CD4+ T cells in the absence of IL-6 trans signaling was largely due to a contraction in type-1 regulatory T cells (CD4+IL10+IFNγ+), with a smaller reduction seen in Tregs (CD4+FoxP3+; Figure 4F-G). Supporting these cellular data, multiple inflammatory cytokines were elevated in the serum early after transplantation when IL-6 trans signaling was inhibited (Figure 4H).
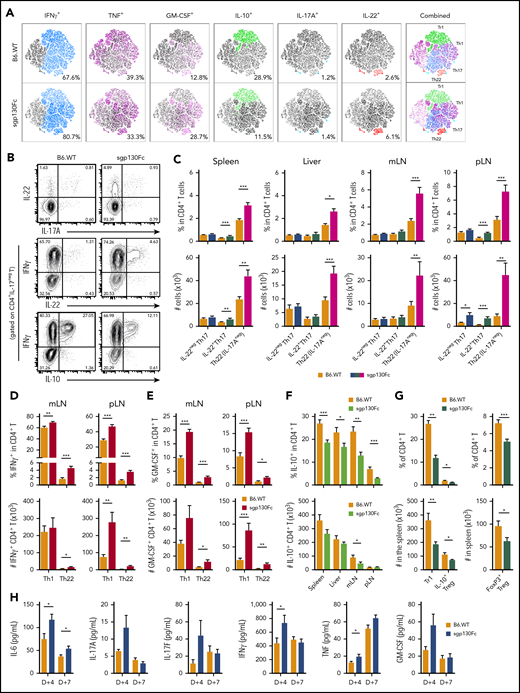
IL-6 trans signaling regulates Th17 and Th22 differentiation. (A-J) Donor BALB/c CD4+ T-cell cytokine expression was assessed at day 7 after transplantation in recipient B6 wild-type (B6.WT) and B6.sgp130Fc (IL-6 trans signaling inhibited) mice. (A) High-dimensional analysis of cytokine expression from donor CD4+ T cells isolated from the mLNs using t-distributed stochastic neighbor embedding (6 parameters: IFNγ, TNF, GM-CSF, IL-10, IL-17A, and IL-22; concatenated from 1 experiment, where n = 6 mice per group). (B) Representative contour plots of IL-22 and IL-17A expression in CD4+ T cells and IFNγ, IL-22, and IL-10 in IL-17A− CD4+ T cells from the mLNs (concatenated from 4-5 mice per group). (C) Proportion and total numbers of Th17 cells (CD4+IL-17A+) either not expressing IL-22 (IL-22−Th17) or expressing IL-22 (IL-22+Th17) and Th22 cells (CD4+IL-17A−IL-22+) in the spleen, liver, mLNs, and peripheral LNs (pLNs) of B6.WT and sgp130Fc mice (n = 10-22 mice per group from 2-4 separate experiments). (D-E) Frequency and total numbers of Th1 (CD4+IFNγ+IL-17A−IL-22−) and Th22 (CD4+IL-17A−IL-22+) cells expressing IFNγ (D) and GM-CSF (E) as a proportion of total CD4+ T cells (n = 10-11 mice per group from 2-3 separate experiments). (F) Frequency and total number of CD4+ T cells expressing IL-10 in multiple organs (10-22 mice per group from 2-4 separate experiments). (G) Percentage and total number of type-1 T regulatory cells (Tregs) (Tr1, CD4+IFNγ+IL-10+) and FoxP3+ Tregs expressing IL-10 (CD4+FoxP3+IL-10+) in the spleen and FoxP3+ Tregs (CD4+FoxP3+) as a percentage of total CD4+ T cells in the spleen (n = 6-13 mice per group from 1-2 experiments). (H) Peripheral blood serum cytokine levels of IL-6, IL-17A, IL-17F, IFNγ, TNF, and GM-CSF at day 4 and day 7 after transplantation (n = 16-28 mice per group from 4 experiments). Data presented as mean ± standard error of the mean. *P < .05, **P < .01, ***P < .001.
IL-6 trans signaling regulates Th17 and Th22 differentiation. (A-J) Donor BALB/c CD4+ T-cell cytokine expression was assessed at day 7 after transplantation in recipient B6 wild-type (B6.WT) and B6.sgp130Fc (IL-6 trans signaling inhibited) mice. (A) High-dimensional analysis of cytokine expression from donor CD4+ T cells isolated from the mLNs using t-distributed stochastic neighbor embedding (6 parameters: IFNγ, TNF, GM-CSF, IL-10, IL-17A, and IL-22; concatenated from 1 experiment, where n = 6 mice per group). (B) Representative contour plots of IL-22 and IL-17A expression in CD4+ T cells and IFNγ, IL-22, and IL-10 in IL-17A− CD4+ T cells from the mLNs (concatenated from 4-5 mice per group). (C) Proportion and total numbers of Th17 cells (CD4+IL-17A+) either not expressing IL-22 (IL-22−Th17) or expressing IL-22 (IL-22+Th17) and Th22 cells (CD4+IL-17A−IL-22+) in the spleen, liver, mLNs, and peripheral LNs (pLNs) of B6.WT and sgp130Fc mice (n = 10-22 mice per group from 2-4 separate experiments). (D-E) Frequency and total numbers of Th1 (CD4+IFNγ+IL-17A−IL-22−) and Th22 (CD4+IL-17A−IL-22+) cells expressing IFNγ (D) and GM-CSF (E) as a proportion of total CD4+ T cells (n = 10-11 mice per group from 2-3 separate experiments). (F) Frequency and total number of CD4+ T cells expressing IL-10 in multiple organs (10-22 mice per group from 2-4 separate experiments). (G) Percentage and total number of type-1 T regulatory cells (Tregs) (Tr1, CD4+IFNγ+IL-10+) and FoxP3+ Tregs expressing IL-10 (CD4+FoxP3+IL-10+) in the spleen and FoxP3+ Tregs (CD4+FoxP3+) as a percentage of total CD4+ T cells in the spleen (n = 6-13 mice per group from 1-2 experiments). (H) Peripheral blood serum cytokine levels of IL-6, IL-17A, IL-17F, IFNγ, TNF, and GM-CSF at day 4 and day 7 after transplantation (n = 16-28 mice per group from 4 experiments). Data presented as mean ± standard error of the mean. *P < .05, **P < .01, ***P < .001.
IL-6 trans signaling regulates GVHD via the suppression of donor Th22 differentiation
We previously reported that donor Th22 cells can induce cutaneous GVHD.29 To investigate whether inhibition of IL-6 trans signaling was driving cGVHD via the expansion of Th22 cells, we transplanted donor grafts deficient in IL-22. Indeed, transplantation of IL-22−/− grafts eliminated the excess cutaneous GVHD and mortality seen in the sgp130Fc transgenic recipients (Figure 5A-D). Furthermore, the excess of Th22 cells seen after inhibition of IL-6 trans signaling was dependent upon IL-6 classical signaling, because this was reversed by either specific deletion of IL-6R on T cells (Figure 5E-G) or blockade of both IL-6 classical and trans pathways after anti–IL-6R antibody administration (Figure 5H-I). Therefore, these data demonstrate that in the absence of IL-6 trans signaling, excess IL-6 classical signaling promotes Th22 differentiation and cutaneous GVHD.
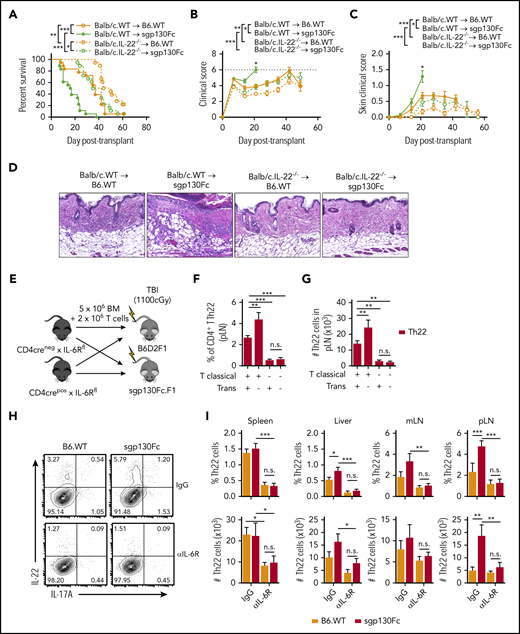
IL-6 trans signaling regulates GVHD via the suppression of donor Th22 differentiation. (A-D) Lethally irradiated B6 wild-type (B6.WT) and sgp130Fc (IL-6 trans signaling inhibited) mice received 10 × 106 BALB/c BM and 5 × 106 T-cell allografts from WT or IL-22–deficient donors (BALB/c.IL-22−/−). (A-C) Survival indices by Kaplan-Meier analyses (A), combined clinical scores (B), and scores of skin pathology (C) after transplantation (n = 18 mice per group from 3 experiments). (D) Representative skin sections taken at day 21 after transplantation (hematoxylin and eosin stain; 200× magnification). (E) Lethally irradiated B6D2F1 and sgp130Fc.F1 (IL-6 trans signaling inhibited) mice received BM and T-cell allografts from control mice (CD4Cre− × IL-6Rfl) or mice with T cells deficient in IL-6 classical signaling (CD4Cre+ × IL-6Rfl) and frequency (F) and total number (G) of Th22 cells measured at day 7 after transplantation in the peripheral LNs (pLNs) (n = 8 mice per group from 2 experiments). (H-J) Recipient B6.WT and sgp130Fc (IL-6 trans signaling inhibited) mice were treated with anti–IL-6R antibody (all IL-6 signaling pathways blocked) or control immunoglobulin G (IgG) antibody and CD4+ T-cell differentiation assessed at day 7 after transplantation of BALB/c.WT T cells. (H-I) Representative contour plots of IL-22 and IL-17A expression in CD4+ T cells from the pLNs (concatenated from 4 mice per group) (H) and frequency of CD4+ T cells and total number of Th22 cells (I) (CD4+IL-22+IL-17A−; n = 8 mice per group from 2 experiments). Data presented as mean ± standard error of the mean. *P < .05, **P < .01, ***P < .001. n.s., not significant; TBI, total-body irradiation.
IL-6 trans signaling regulates GVHD via the suppression of donor Th22 differentiation. (A-D) Lethally irradiated B6 wild-type (B6.WT) and sgp130Fc (IL-6 trans signaling inhibited) mice received 10 × 106 BALB/c BM and 5 × 106 T-cell allografts from WT or IL-22–deficient donors (BALB/c.IL-22−/−). (A-C) Survival indices by Kaplan-Meier analyses (A), combined clinical scores (B), and scores of skin pathology (C) after transplantation (n = 18 mice per group from 3 experiments). (D) Representative skin sections taken at day 21 after transplantation (hematoxylin and eosin stain; 200× magnification). (E) Lethally irradiated B6D2F1 and sgp130Fc.F1 (IL-6 trans signaling inhibited) mice received BM and T-cell allografts from control mice (CD4Cre− × IL-6Rfl) or mice with T cells deficient in IL-6 classical signaling (CD4Cre+ × IL-6Rfl) and frequency (F) and total number (G) of Th22 cells measured at day 7 after transplantation in the peripheral LNs (pLNs) (n = 8 mice per group from 2 experiments). (H-J) Recipient B6.WT and sgp130Fc (IL-6 trans signaling inhibited) mice were treated with anti–IL-6R antibody (all IL-6 signaling pathways blocked) or control immunoglobulin G (IgG) antibody and CD4+ T-cell differentiation assessed at day 7 after transplantation of BALB/c.WT T cells. (H-I) Representative contour plots of IL-22 and IL-17A expression in CD4+ T cells from the pLNs (concatenated from 4 mice per group) (H) and frequency of CD4+ T cells and total number of Th22 cells (I) (CD4+IL-22+IL-17A−; n = 8 mice per group from 2 experiments). Data presented as mean ± standard error of the mean. *P < .05, **P < .01, ***P < .001. n.s., not significant; TBI, total-body irradiation.
IL-6 classical signaling in donor T cells drives Th22- and Th17-dependent GVHD
Given the regulatory function of IL-6 trans signaling after alloBMT, we next investigated the role of IL-6 classical signaling in GVHD. To this end, IL-6Rfl was crossed with CD4Cre+ mice, removing IL-6R required for classical signaling but leaving gp130 expression intact, facilitating trans signaling (Figure 6A). CD4+ T cells in CD4Cre+ × IL-6Rfl mice lacked membrane-bound IL-6R expression and were unable to upregulate phosphorylated STAT3 in response to IL-6 stimulation; however, they were still responsive to IL-6/sIL-6R trans signaling complexes (hyper–IL-647 ; Figure 6B-C). After transplantation, deletion of IL-6R expression on donor T cells significantly protected mice from lethal aGVHD (Figure 6D). Moreover, this effect was specific to donor CD4+ T cells, because IL-6R–deficient CD8+ T cells induced significant GVHD (Figure 6E). The improved survival was not associated with differences in the Th1 cytokine IFNγ, TNF, or GM-CSF (Figure 6F-G); however, both Th22 and Th17 populations were critically dependent upon IL-6R expression for their differentiation (Figure 6H-I). Supporting these data, the decrease of Th22 and Th17 cells was corroborated by reduced serum IL-17A levels and expression of their transcription factor RORγt in CD4+ T cells, with a concurrent increase in FoxP3+ Tregs (Figure 6J-M). Therefore, IL-6 classical signaling acts on donor CD4+ T cells to drive GVHD mortality via Th17 and Th22 differentiation, while simultaneously suppressing Treg expansion.
![IL-6 classical signaling in donor T cells drives Th22- and Th17-dependent GVHD. (A-L) Lethally irradiated (1300 cGy) B6D2F1 mice received BM and T cells or TCD BM allografts from control mice (CD4Cre− × IL-6Rfl) or mice with T cells deficient in IL-6 classical signaling (CD4Cre+ × IL-6Rfl). (B) Deletion of IL-6R on Cre+ recipients was confirmed by flow cytometric analysis of mIL-6R expression on peripheral blood CD4+ T cells before transplantation. (C) Functional confirmation was performed by stimulation of CD4+ T cells (in peripheral whole blood) with IL-6 (to induce classical signaling) or hyper–IL-6 (H-IL-6; to induce trans signaling) and upregulation of phosphorylated STAT3 (pSTAT3) expression measured by flow cytometry. (D) Survival indices by Kaplan-Meier analyses and weekly clinical GVHD scores of recipients after transplantation (BM and T-cell grafts, n = 12 mice per group; TCD, n = 6 mice per group; from 2 experiments). (E) Survival indices of recipient mice receiving grafts containing BM and the listed combination of sorted CD4+ (CD90.2+CD4+γδTCR−7AAD−) or CD8+ (CD90.2+CD4−γδTCR−7AAD−) T cells from CD4Cre− × IL-6Rfl (CD4 wild-type [CD4.WT] or CD8.WT) or CD4Cre+ × IL-6Rfl mice (T-cell grafts, n = 24 mice per group; TCD grafts, n = 8 mice per group; from 4 experiments). (F-I) CD4+ T-cell cytokine expression was assessed at day 7 after transplantation after stimulation. (F-G) Representative contour plots (mLNs; concatenated from 5 mice per group) (F) and frequencies of IFNγ, TNF, and GM-CSF expressing in CD4+ T cells isolated from the spleen and mLNs (n = 10 mice per group from 2 experiments) (G). (H-I) Representative flow cytometry plots of IL-22 and IL-17A in CD4+ T cells isolated from the mLNs (concatenated from 5 mice per group) (H) and frequencies and total numbers of Th22 (CD4+IL-22+IL-17−), IL-22+Th17 (CD4+IL-22+IL-17+), and IL-22−Th17 (CD4+IL-22−IL-17+) in the spleen, liver, mLNs, and peripheral LNs (pLNs) (n = 10 mice per group from 2 experiments) (I). (J) Peripheral blood serum levels of IL-17A at day 4 and day 7 after transplantation. (K-M) Frequency and total numbers of Tbet+ and RORγt+ in CD4+ T cells (K), Tr1 cells (CD4+IFNγ+IL-10+EOMES+) (L),73 and Tregs (CD4+FoxP3+) in the spleen at day 7 after transplantation (M) (n = 10 mice per group from 2 experiments). Data presented as mean ± standard error of the mean. *P < .05, **P < .01, ***P < .001. TBI, total-body irradiation.](https://ash.silverchair-cdn.com/ash/content_public/journal/blood/134/23/10.1182_blood.2019000396/4/m_bloodbld2019000396f6.png?Expires=1769082003&Signature=V6H~mgXKJNLmlDxsO76RZDLmlzIamgWsZEtlCagZjKVJO1f5Q0BaWjyBodXxEsoancQavoEP0uwefM~sZJ9MoXCtoSHvxhY0rl3N1IVV8kjjCq3-SwV8y~4CLVd5H0H4PRibPAiLxSHYJ41pXElYRYhFEq9roAA-9r7RdtXS6INQK~zGnT-Vp0p9MrUIm8aPTw6h4zYINCA2D4jLJgItZ4ltS0zvJKaA4mUL1bimURjDYK0e1-6NxtQZ2HnGNTfb9wpP5-~xGv9KbUazla5A5~OxP~xUQhIYviaNHKIoGrQsWc0o1DBFh4f2N5sUezIUTNxTsn0hozZGgR92-imZQA__&Key-Pair-Id=APKAIE5G5CRDK6RD3PGA)
IL-6 classical signaling in donor T cells drives Th22- and Th17-dependent GVHD. (A-L) Lethally irradiated (1300 cGy) B6D2F1 mice received BM and T cells or TCD BM allografts from control mice (CD4Cre− × IL-6Rfl) or mice with T cells deficient in IL-6 classical signaling (CD4Cre+ × IL-6Rfl). (B) Deletion of IL-6R on Cre+ recipients was confirmed by flow cytometric analysis of mIL-6R expression on peripheral blood CD4+ T cells before transplantation. (C) Functional confirmation was performed by stimulation of CD4+ T cells (in peripheral whole blood) with IL-6 (to induce classical signaling) or hyper–IL-6 (H-IL-6; to induce trans signaling) and upregulation of phosphorylated STAT3 (pSTAT3) expression measured by flow cytometry. (D) Survival indices by Kaplan-Meier analyses and weekly clinical GVHD scores of recipients after transplantation (BM and T-cell grafts, n = 12 mice per group; TCD, n = 6 mice per group; from 2 experiments). (E) Survival indices of recipient mice receiving grafts containing BM and the listed combination of sorted CD4+ (CD90.2+CD4+γδTCR−7AAD−) or CD8+ (CD90.2+CD4−γδTCR−7AAD−) T cells from CD4Cre− × IL-6Rfl (CD4 wild-type [CD4.WT] or CD8.WT) or CD4Cre+ × IL-6Rfl mice (T-cell grafts, n = 24 mice per group; TCD grafts, n = 8 mice per group; from 4 experiments). (F-I) CD4+ T-cell cytokine expression was assessed at day 7 after transplantation after stimulation. (F-G) Representative contour plots (mLNs; concatenated from 5 mice per group) (F) and frequencies of IFNγ, TNF, and GM-CSF expressing in CD4+ T cells isolated from the spleen and mLNs (n = 10 mice per group from 2 experiments) (G). (H-I) Representative flow cytometry plots of IL-22 and IL-17A in CD4+ T cells isolated from the mLNs (concatenated from 5 mice per group) (H) and frequencies and total numbers of Th22 (CD4+IL-22+IL-17−), IL-22+Th17 (CD4+IL-22+IL-17+), and IL-22−Th17 (CD4+IL-22−IL-17+) in the spleen, liver, mLNs, and peripheral LNs (pLNs) (n = 10 mice per group from 2 experiments) (I). (J) Peripheral blood serum levels of IL-17A at day 4 and day 7 after transplantation. (K-M) Frequency and total numbers of Tbet+ and RORγt+ in CD4+ T cells (K), Tr1 cells (CD4+IFNγ+IL-10+EOMES+) (L),73 and Tregs (CD4+FoxP3+) in the spleen at day 7 after transplantation (M) (n = 10 mice per group from 2 experiments). Data presented as mean ± standard error of the mean. *P < .05, **P < .01, ***P < .001. TBI, total-body irradiation.
IL-6 classical signaling in donor T cells drives Th22- and Th17-dependent GVHD. (A-L) Lethally irradiated (1300 cGy) B6D2F1 mice received BM and T cells or TCD BM allografts from control mice (CD4Cre− × IL-6Rfl) or mice with T cells deficient in IL-6 classical signaling (CD4Cre+ × IL-6Rfl). (B) Deletion of IL-6R on Cre+ recipients was confirmed by flow cytometric analysis of mIL-6R expression on peripheral blood CD4+ T cells before transplantation. (C) Functional confirmation was performed by stimulation of CD4+ T cells (in peripheral whole blood) with IL-6 (to induce classical signaling) or hyper–IL-6 (H-IL-6; to induce trans signaling) and upregulation of phosphorylated STAT3 (pSTAT3) expression measured by flow cytometry. (D) Survival indices by Kaplan-Meier analyses and weekly clinical GVHD scores of recipients after transplantation (BM and T-cell grafts, n = 12 mice per group; TCD, n = 6 mice per group; from 2 experiments). (E) Survival indices of recipient mice receiving grafts containing BM and the listed combination of sorted CD4+ (CD90.2+CD4+γδTCR−7AAD−) or CD8+ (CD90.2+CD4−γδTCR−7AAD−) T cells from CD4Cre− × IL-6Rfl (CD4 wild-type [CD4.WT] or CD8.WT) or CD4Cre+ × IL-6Rfl mice (T-cell grafts, n = 24 mice per group; TCD grafts, n = 8 mice per group; from 4 experiments). (F-I) CD4+ T-cell cytokine expression was assessed at day 7 after transplantation after stimulation. (F-G) Representative contour plots (mLNs; concatenated from 5 mice per group) (F) and frequencies of IFNγ, TNF, and GM-CSF expressing in CD4+ T cells isolated from the spleen and mLNs (n = 10 mice per group from 2 experiments) (G). (H-I) Representative flow cytometry plots of IL-22 and IL-17A in CD4+ T cells isolated from the mLNs (concatenated from 5 mice per group) (H) and frequencies and total numbers of Th22 (CD4+IL-22+IL-17−), IL-22+Th17 (CD4+IL-22+IL-17+), and IL-22−Th17 (CD4+IL-22−IL-17+) in the spleen, liver, mLNs, and peripheral LNs (pLNs) (n = 10 mice per group from 2 experiments) (I). (J) Peripheral blood serum levels of IL-17A at day 4 and day 7 after transplantation. (K-M) Frequency and total numbers of Tbet+ and RORγt+ in CD4+ T cells (K), Tr1 cells (CD4+IFNγ+IL-10+EOMES+) (L),73 and Tregs (CD4+FoxP3+) in the spleen at day 7 after transplantation (M) (n = 10 mice per group from 2 experiments). Data presented as mean ± standard error of the mean. *P < .05, **P < .01, ***P < .001. TBI, total-body irradiation.
IL-6R expression in the GI tract is dispensable for GVHD
We previously reported that IL-6 may induce direct cytotoxic damage to host intestinal epithelial cells in vitro,22 although IL-6 is also an important mediator of intestinal regeneration.15,48-53 Therefore, we next investigated the role of IL-6 classical signaling regarding intestinal cells in vivo by using tamoxifen-inducible Villin-creERT × IL-6Rfl mice, removing IL-6R expression on intestinal epithelial and stem cells.33,34 Similar to blockade of IL-6 trans signaling within the gut (Figure 3E-I), the absence of IL-6 classical signaling in intestinal epithelia did not influence aGVHD pathology in the GI tract or mortality (Figure 7A-C). Therefore, IL-6 is unlikely to mediate important protective or pathogenic GVHD processes in the GI tract via direct effects on the epithelia.
![IL-6 signaling is dispensable for GI tract pathology and GVL responses. (A-C) Villin-Cre− × IL-6Rfl and Villin-Cre+ (Villin-CreERT2) × IL-6Rfl mice were treated with tamoxifen to induce Cre recombinase activity and cohoused. Two weeks after the first tamoxifen injection, recipient mice were lethally irradiated and received allografts containing 10 × 106 BM and 3 × 106 to 5 × 106 T cells or 10 × 106 TCD BM alone from BALB/c.45.1 donors. (A) Survival indices and weight loss percentage of recipients after transplantation (T-cell grafts, n = 14 mice per group from 3 experiments; TCD, n = 2-3 mice from 1 experiment). (B-C) Representative histology sections of ileum at day 6 to 7 after transplantation (200× magnification) (B) and semiquantitative GVHD histopathology (C) (n = 8-9 mice per group from 2 experiments). (D-F) The direct responsiveness of leukemic cells to IL-6 was first assessed. Lethally irradiated B6D2F1 mice received B6 wild-type (B6.WT) TCD BM grafts supplemented with either 1 × 106 B6D2F1-derived BCR-ABL/NUP98-HOXA9 GFP+ chronic myeloid leukemia blast crisis or MLL-AF9 GFP+ acute myeloid leukemia cells. (D-E) Peripheral blood was collected from mice after engraftment and expression of mIL-6R (D) and gp130 (E) determined on GFP+ cells. (F) The functional ability of the leukemia cells to respond to IL-6 was confirmed by whole-blood stimulation with IL-6 (to induce classical signaling) or hyper–IL-6 (H-IL-6) (to induce trans signaling) and upregulation of phosphorylated STAT3 (pSTAT3) measured by flow cytometry. (G-I) To assess the role of donor IL-6 classical signaling on GVL responses, lethally irradiated B6D2F1 mice received grafts containing 1 × 106 BCR-ABL/NUP98-HOXA9 GFP+ leukemic cells along with either TCD BM and T cells (0.1 × 106) or TCD BM alone (non-GVL control) from either VAVCre− × IL-6Rfl (control) or VAVCre+ × IL-6Rfl mice (donor leukocytes deficient in IL-6 classical signaling) (G); survival indices by Kaplan-Meier analyses (H) and peripheral blood leukemia burden (I) at the indicated day after transplantation (survival indices, n = 8-12 mice per group; percentage leukocyte GFP+, n = 4-12 mice per group; from 2 experiments). (J-K) To assess the role of IL-6 trans signaling on GVL responses, lethally irradiated B6D2F1 (control) or sgp130Fc.F1 (IL-6 trans signaling inhibited) mice received grafts containing MLL-AF9 GFP+ cells with TCD BM and T cells (0.5 × 106 T cells [T]) or TCD BM alone (non-GVL control) from B6.WT donor mice. (J) Survival indices by Kaplan-Meier analysis (n = 11 mice per group from 2 experiments). (K) Representative images of blood smears at day 21 after transplantation (Wright-Giemsa stain; 200× magnification). Data presented as mean ± standard error of the mean. *P < .05, **P < .01, ***P < .001. TBI, total-body irradiation.](https://ash.silverchair-cdn.com/ash/content_public/journal/blood/134/23/10.1182_blood.2019000396/4/m_bloodbld2019000396f7.png?Expires=1769082003&Signature=Xhj-Tv01iZk1awUZVCqMYXgb20pDLGbKJmJ1CDI3~~6KjYcSGvyNnd7DHU2sjrngtiBs~qkCC5qbNjOik8aKN-HhqWApNdu4SrKm04l8EVu7ZUJp~uPhy1Be5PHxwldyPuKkfg5eqBxUlyS0i0wLvQNEDDmg6w~Dlhx9PISTT7L78DPjHPLuZe4cgRqsWNVFYaxM6BDEjAoE2Sh0ddfFh7j9kMaezlCkN7roF4oRok4Mip3CysZPQ5iWQ2~eu4j3Z4DDJUynzVq6kJ1JlG6uzR8f7XPyHGHSfj7w928bU-8cR79re2jBgRnNiwTq2msfTMEmInXwmaPPNd7gmMu4oA__&Key-Pair-Id=APKAIE5G5CRDK6RD3PGA)
IL-6 signaling is dispensable for GI tract pathology and GVL responses. (A-C) Villin-Cre− × IL-6Rfl and Villin-Cre+ (Villin-CreERT2) × IL-6Rfl mice were treated with tamoxifen to induce Cre recombinase activity and cohoused. Two weeks after the first tamoxifen injection, recipient mice were lethally irradiated and received allografts containing 10 × 106 BM and 3 × 106 to 5 × 106 T cells or 10 × 106 TCD BM alone from BALB/c.45.1 donors. (A) Survival indices and weight loss percentage of recipients after transplantation (T-cell grafts, n = 14 mice per group from 3 experiments; TCD, n = 2-3 mice from 1 experiment). (B-C) Representative histology sections of ileum at day 6 to 7 after transplantation (200× magnification) (B) and semiquantitative GVHD histopathology (C) (n = 8-9 mice per group from 2 experiments). (D-F) The direct responsiveness of leukemic cells to IL-6 was first assessed. Lethally irradiated B6D2F1 mice received B6 wild-type (B6.WT) TCD BM grafts supplemented with either 1 × 106 B6D2F1-derived BCR-ABL/NUP98-HOXA9 GFP+ chronic myeloid leukemia blast crisis or MLL-AF9 GFP+ acute myeloid leukemia cells. (D-E) Peripheral blood was collected from mice after engraftment and expression of mIL-6R (D) and gp130 (E) determined on GFP+ cells. (F) The functional ability of the leukemia cells to respond to IL-6 was confirmed by whole-blood stimulation with IL-6 (to induce classical signaling) or hyper–IL-6 (H-IL-6) (to induce trans signaling) and upregulation of phosphorylated STAT3 (pSTAT3) measured by flow cytometry. (G-I) To assess the role of donor IL-6 classical signaling on GVL responses, lethally irradiated B6D2F1 mice received grafts containing 1 × 106 BCR-ABL/NUP98-HOXA9 GFP+ leukemic cells along with either TCD BM and T cells (0.1 × 106) or TCD BM alone (non-GVL control) from either VAVCre− × IL-6Rfl (control) or VAVCre+ × IL-6Rfl mice (donor leukocytes deficient in IL-6 classical signaling) (G); survival indices by Kaplan-Meier analyses (H) and peripheral blood leukemia burden (I) at the indicated day after transplantation (survival indices, n = 8-12 mice per group; percentage leukocyte GFP+, n = 4-12 mice per group; from 2 experiments). (J-K) To assess the role of IL-6 trans signaling on GVL responses, lethally irradiated B6D2F1 (control) or sgp130Fc.F1 (IL-6 trans signaling inhibited) mice received grafts containing MLL-AF9 GFP+ cells with TCD BM and T cells (0.5 × 106 T cells [T]) or TCD BM alone (non-GVL control) from B6.WT donor mice. (J) Survival indices by Kaplan-Meier analysis (n = 11 mice per group from 2 experiments). (K) Representative images of blood smears at day 21 after transplantation (Wright-Giemsa stain; 200× magnification). Data presented as mean ± standard error of the mean. *P < .05, **P < .01, ***P < .001. TBI, total-body irradiation.
IL-6 signaling is dispensable for GI tract pathology and GVL responses. (A-C) Villin-Cre− × IL-6Rfl and Villin-Cre+ (Villin-CreERT2) × IL-6Rfl mice were treated with tamoxifen to induce Cre recombinase activity and cohoused. Two weeks after the first tamoxifen injection, recipient mice were lethally irradiated and received allografts containing 10 × 106 BM and 3 × 106 to 5 × 106 T cells or 10 × 106 TCD BM alone from BALB/c.45.1 donors. (A) Survival indices and weight loss percentage of recipients after transplantation (T-cell grafts, n = 14 mice per group from 3 experiments; TCD, n = 2-3 mice from 1 experiment). (B-C) Representative histology sections of ileum at day 6 to 7 after transplantation (200× magnification) (B) and semiquantitative GVHD histopathology (C) (n = 8-9 mice per group from 2 experiments). (D-F) The direct responsiveness of leukemic cells to IL-6 was first assessed. Lethally irradiated B6D2F1 mice received B6 wild-type (B6.WT) TCD BM grafts supplemented with either 1 × 106 B6D2F1-derived BCR-ABL/NUP98-HOXA9 GFP+ chronic myeloid leukemia blast crisis or MLL-AF9 GFP+ acute myeloid leukemia cells. (D-E) Peripheral blood was collected from mice after engraftment and expression of mIL-6R (D) and gp130 (E) determined on GFP+ cells. (F) The functional ability of the leukemia cells to respond to IL-6 was confirmed by whole-blood stimulation with IL-6 (to induce classical signaling) or hyper–IL-6 (H-IL-6) (to induce trans signaling) and upregulation of phosphorylated STAT3 (pSTAT3) measured by flow cytometry. (G-I) To assess the role of donor IL-6 classical signaling on GVL responses, lethally irradiated B6D2F1 mice received grafts containing 1 × 106 BCR-ABL/NUP98-HOXA9 GFP+ leukemic cells along with either TCD BM and T cells (0.1 × 106) or TCD BM alone (non-GVL control) from either VAVCre− × IL-6Rfl (control) or VAVCre+ × IL-6Rfl mice (donor leukocytes deficient in IL-6 classical signaling) (G); survival indices by Kaplan-Meier analyses (H) and peripheral blood leukemia burden (I) at the indicated day after transplantation (survival indices, n = 8-12 mice per group; percentage leukocyte GFP+, n = 4-12 mice per group; from 2 experiments). (J-K) To assess the role of IL-6 trans signaling on GVL responses, lethally irradiated B6D2F1 (control) or sgp130Fc.F1 (IL-6 trans signaling inhibited) mice received grafts containing MLL-AF9 GFP+ cells with TCD BM and T cells (0.5 × 106 T cells [T]) or TCD BM alone (non-GVL control) from B6.WT donor mice. (J) Survival indices by Kaplan-Meier analysis (n = 11 mice per group from 2 experiments). (K) Representative images of blood smears at day 21 after transplantation (Wright-Giemsa stain; 200× magnification). Data presented as mean ± standard error of the mean. *P < .05, **P < .01, ***P < .001. TBI, total-body irradiation.
IL-6 is dispensable for GVL responses
The quality of GVL responses after alloBMT correlates with GVHD.54,55 Because GVHD is significantly attenuated after IL-6 inhibition, we next investigated whether this also had a negative impact on GVL responses. To separate the role of IL-6 on the donor graft and direct signaling to leukemia cells, we used 2 myeloid leukemia cell lines that were either directly unresponsive or responsive to IL-6: BCR-ABL/NUP98-HOXA9, a primary blast crisis chronic myeloid leukemia35 that did not express IL-6R or gp130 and was subsequently unresponsive to IL-6 (Figure 7D-F), and the MLL-AF9 acute myeloid leukemia cell line36 that expressed low levels of IL-6R and gp130 and was responsive to IL-6 (Figure 7D-F).
Recipient mice were lethally irradiated and received grafts containing leukemia and TCD BM alone (non-GVL) or T cells to induce GVL responses (Figure 7G). The T-cell doses used in these models were insufficient to drive lethal GVHD, and thus, mortality represents leukemia deaths only. We eliminated all IL-6 classical signaling by transplanting grafts where IL-6R was deleted by VAV-driven Cre recombinase. Despite significant attenuation of GVHD mortality after the elimination of IL-6 classical signaling in T cells (Figure 6D), here in the context of GVL responses, we did not observe a significant difference in GVL to BCR-ABL/NUP98-HOXA9 chronic myeloid leukemia (Figure 7H-I). We observed a modest prolongation in survival in mice receiving MLL-AF9 acute myeloid leukemia cells in the presence of IL-6 trans signaling inhibition (Figure 7J-K). However, this effect was also evident in the absence of donor T cells. Because MLL-AF9 cells are directly responsive to IL-6, this suggests that IL-6 trans signaling directly enhances MLL-AF9 leukemia growth, likely via augmentation of classical IL-6 signaling in leukemia. Therefore, collectively, IL-6 classical signaling in donor cells attenuates GVHD but does not significantly influence GVL responses.
Discussion
In this study, we delineate the complex mechanisms involved in IL-6–driven GVHD by identifying the source of IL-6, the signaling pathways involved, and its cellular targets. Here we demonstrate that, although IL-6 is expressed by most hematopoietic and nonhematopoietic cells, systemic dysregulation of IL-6 after transplantation is critically dependent upon recipient DCs. DC-derived IL-6 was then required for downstream differentiation of donor CD4+ T cells into polyinflammatory Th17 and Th22 subsets. We further confirm that this process is directed through the IL-6 classical pathway on CD4+ T cells, orchestrating Th17 and Th22 development, impeding Treg formation, and inducing fulminant GVHD. Similarly, after donor engraftment, donor DCs then assume this role, providing IL-6 to donor T cells to drive their expansion and differentiation. Thus, here we demonstrate that IL-6 classical signaling in the donor graft, driven by DC-derived IL-6, is a key mediator of GVHD.
Clinical inhibition of IL-6 or IL-6R is an effective strategy for many inflammatory disorders; however, recent evidence suggests that similar efficacy may be achieved by selective blockade of the IL-6 trans pathway, potentially maintaining some of the protective processes of IL-6.56 Interestingly, here we show that after alloBMT, IL-6 trans signaling was not required for CD4+ T-cell differentiation or their migration to target organs, but it instead suppressed their systemic development into pathogenic and polyinflammatory donor Th22 cells. Supporting our previous findings, these Th22 cells coexpressed IFNγ, TNF, and GM-CSF and were capable of driving severe, IL-22–dependent cutaneous cGVHD.29 Therefore, these data warn against clinical approaches to block IL-6 trans signaling after alloSCT with sgp130Fc (Olamkicept).44,57 Importantly however, although specific blockade of IL-6 trans signaling alone exacerbated GVHD, we further show that this process was driven by the classical pathway and was subsequently reversible by additional IL-6R inhibition. Therefore, although the anti–IL-6R antibody inhibits both IL-6 classical and trans signaling, blockade of both pathways is unlikely to exacerbate clinical GVHD.
In other inflammatory disease models, IL-6–driven pathology was primarily mediated through the trans signaling pathway, whereby pathology could be attenuated with sgp130Fc treatment.15,58-61 In rheumatoid arthritis models, this was associated with IL-6 trans signaling–mediated leukocyte trafficking, driving cellular infiltration (including Th17 cells) into localized sites of inflammation.58,59 Although consistent with our findings, it was later reported that pathogenic T cells required the classical pathway for systemic disease.60 Because T-cell IL-6R expression is lost after activation, IL-6 trans signaling has also been proposed to be required for signaling on activated T cells via ligation to gp130.62,63 This premise was further supported in an experimental colitis model, where IL-6 trans signaling prevented T-cell apoptosis in the gut, and later, trans signaling was shown to provide Th17 maintenance after their induction.61,64 However, after alloBMT, we did not observe a significant role for trans signaling in Th17 development, maintenance, or infiltration into multiple GVHD target organs. These data likely reflect the downregulation of gp130 on T cells after alloSCT,37 suggesting that after activation or IL-6 signaling, T cells downregulate both IL-6R and gp130, rendering them refractory to further IL-6 signaling. Another difference between alloBMT and other inflammatory disorders is the induction of pathogenic Th17 cells by cognate, transpresentation of IL-6/IL-6R complexes by DCs (cluster signaling), described in an experimental autoimmune encephalomyelitis model.17 Here we did not observe an important role for IL-6R expression on DCs, and therefore for cluster signaling, in the induction of polyinflammatory Th17 cells. Collectively, these data suggest that IL-6–induced pathology is highly contextual and thus requires disease-specific analysis to inform rational approaches to clinical inhibition.
Because the main therapeutic potential of alloBMT stems from the GVL effect, and GVHD is a consequence of this alloreactivity, isolating these competing effects is difficult but highly desirable for clinical therapies. Here we report that although removing IL-6 classical signaling on T cells reduced GVHD mortality, it did not clearly influence the ability of the donor graft to protect from leukemic relapse. Concordant with these findings, both donor Th17 and Th22 cells are dispensable for donor GVL responses and are dependent on IL-6 classical signaling.22,30,65,66 Therefore, these data suggest that coblockade of both classical and trans pathways with anti–IL-6R treatment may reduce GVHD without compromising GVL responses. A caveat of these studies is that the leukemias used generally do not express major histocompatibility complex class 2, and so these studies should be confirmed in major histocompatibility complex class 2–expressing malignancies, such as B-cell acute lymphoblastic leukemia. Furthermore, very large clinical data sets will be required to confirm such effects.
One of the more concerning adverse effects associated with IL-6 inhibition in the clinic is the increased incidence of GI complications,14,15,48 which seem to be more severe in inflammatory bowel disease patients.49,50 Because of these complications, specific blockade of the IL-6 trans signaling pathway has been proposed as a superior strategy in inflammatory bowel disease to inhibit the inflammatory properties of IL-6 while maintaining its protective processes.15 Supporting this notion, several studies have highlighted an important role for IL-6 classical signaling in driving GI regeneration.51,53,67-70 In the context of transplantation, here we did not observe an important protective role for IL-6 trans or classical signaling in the GI system after alloSCT. Furthermore, prophylactic IL-6R blockade after clinical alloSCT did not reveal any significant adverse reactions in the GI tract.11,12 This phenomenon likely reflects the redundancy of other cytokines in GI tract regeneration, such as IL-22,71 but interestingly suggests relatively few GI-targeted consequences of IL-6 inhibition after transplantation. In sum, these data highlight the IL-6 classical signaling pathway as a potential therapeutic target after alloSCT.
For original data, please contact kate.gartlan@qimrberghofer.edu.au or grhill@fredhutch.org.
The online version of this article contains a data supplement.
The publication costs of this article were defrayed in part by page charge payment. Therefore, and solely to indicate this fact, this article is hereby marked “advertisement” in accordance with 18 USC section 1734.
Acknowledgments
The authors acknowledge the assistance of the QIMR Berghofer Flow Cytometry & Imaging Facility, Paula Hall, Michael Rist, and Grace Chojnowski as well as illustrations by Madeleine Flynn. The visual abstract is a modification of an illustration from Hill and Ferrara.72
This work was supported by research grants from the National Health and Medical Research Council and the Ministry of Economy and Competitiveness and European Regional Development Fund SAF2014-56546-R and RTI2018-101105-B-I00. G.R.H. is an Andy Hill CARE Distinguished Researcher. The work of S.R.-J. was funded by grants from the Deutsche Forschungsgemeinschaft (Bonn, Germany; SFB877 project A1) and by the Cluster of Excellence Inflammation at Interfaces.
Authorship
Contribution: A.N.W. designed and performed experiments and wrote the manuscript; K.C., R.D.K., A.S.H., S.A.M., and K.S.E. performed experiments; A.D.C. performed blinded histological assessment; J.H. and S.R.-J. provided essential reagents; P.Z., M.K., A.V., and S.V. designed experiments; K.H.G. designed and performed experiments and helped write the manuscript; and G.R.H. designed experiments and helped write the manuscript.
Conflict-of-interest disclosure: G.R.H. has received funding from Roche for clinical trials of IL-6 inhibition in transplantation. S.R.-J. has acted as a consultant and speaker for AbbVie, Chugai, Genentech, Roche, Pfizer, and Sanofi, declares he is an inventor on patents owned by CONARIS Research Institute, which develops the sgp130Fc protein Olamkicept, together with the company I-Mab, and has stock ownership in CONARIS. The remaining authors declare no competing financial interests.
Correspondence: Geoffrey R. Hill, Fred Hutchinson Cancer Research Center, 1100 Fairview Ave N, Seattle, WA 98109; e-mail: grhill@fredhutch.org.

This feature is available to Subscribers Only
Sign In or Create an Account Close Modal