

In this issue of Blood, report on their investigation of the molecular interaction between mutations affecting DNMT3A and NPM1, and internal tandem duplications (ITDs) of FLT3 in acute myeloid leukemia (AML). They found that when the 3 mutations cooccur in AML, they synergize to drive increased expression of hepatic leukemia factor (HLF), a transcription factor, and are associated with particular characteristics such as increased leukemic stem cell (LSC) frequency and an aberrant immunophenotype (GPR56highCD34low), which seem to be at least in part attributable to overexpression of HLF. Their findings establish HLF overexpression as an important mediator of the adverse phenotype and propose that HLF or its downstream effectors represent potential therapeutic vulnerabilities of this poor-prognosis AML subtype.1
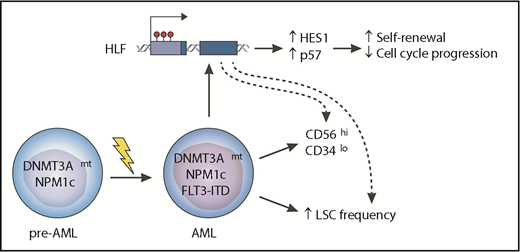
The cooccurrence of oncogenic mutations affecting the DNMT3A and NPM1 genes, together with ITDs in FLT3, is one of the most common combinations of genetic events in AML and seems sufficient to generate the disease. The reasons for why there is a relatively high frequency of this cooccurrence and why it is associated with a worse prognosis for patients are poorly understood. Garg et al report that the 3 mutations synergize to upregulate the HLF gene in association with hypomethylation of its promoter. In turn, HLF activates a set of genes that includes HES1 (a known NOTCH target) and the cell cycle inhibitor p57 (CDKN1C). These changes are associated with enhanced self-renewal of AML cells, an increased LSC frequency, and an aberrant immunophenotype (CD56highCD34low) that is characteristic of the triple-mutant cells. Genetic targeting of HLF sensitized AML cells to antileukemic drugs, suggesting that targeting this pathway may represent a novel therapeutic strategy in this and potentially other AML subtypes.
The cooccurrence of oncogenic mutations affecting the DNMT3A and NPM1 genes, together with ITDs in FLT3, is one of the most common combinations of genetic events in AML and seems sufficient to generate the disease. The reasons for why there is a relatively high frequency of this cooccurrence and why it is associated with a worse prognosis for patients are poorly understood. Garg et al report that the 3 mutations synergize to upregulate the HLF gene in association with hypomethylation of its promoter. In turn, HLF activates a set of genes that includes HES1 (a known NOTCH target) and the cell cycle inhibitor p57 (CDKN1C). These changes are associated with enhanced self-renewal of AML cells, an increased LSC frequency, and an aberrant immunophenotype (CD56highCD34low) that is characteristic of the triple-mutant cells. Genetic targeting of HLF sensitized AML cells to antileukemic drugs, suggesting that targeting this pathway may represent a novel therapeutic strategy in this and potentially other AML subtypes.
Large-scale sequencing studies have revealed the considerable genetic heterogeneity of AML and have identified recurrent mutations in more than 75 genes and chromosomal regions.2,3 Within this complex landscape, patterns of significant cooccurrence and mutual exclusivity allude to mechanistic interactions between different mutations and help define genetic subtypes of AML. Perhaps the most striking such pattern is the 3-way cooccurrence of mutations affecting DNMT3A (particularly at the R882 codon), NPM1 (cytoplasmic mutants, NPM1c), and FLT3 (predominantly FLT3-ITD). The pattern is seen in 6% to 8% of all cases of AML,2,3 a strikingly high percentage for a 3-gene comutation, yet little is known about its precise molecular underpinnings. Furthermore, clinical outcome data show that DNMT3A/NPM1/FLT3-ITD triple-mutant AML has a particularly bad prognosis with a long-term survival below 20%,2 the basis for which is not understood.
To provide insights into these observations, Garg et al initially studied surface expression of the LSC marker GPR56 (which they identified previously4 ) on these AML cells and found that the aberrant GPR56highCD34low immunophenotype was specific to triple-mutant leukemic cells and was not seen in normal hematopoietic stem and progenitor cells (HSPCs) or in AML subclones that did not harbor all 3 mutations. They proceeded to study the transcriptome of triple-mutant AML cells using RNA sequencing and, after focusing specifically on differentially expressed transcription factors, they identified HLF overexpression as a characteristic feature of these leukemias. HLF was originally identified as a partner in the E2A-HLF fusion gene in pediatric acute lymphoblastic leukemia,5 and it also has important roles in normal hematopoiesis,6,7 but it has not been reported to have a role in AML. By examining publically available CpG methylation data sets in AML, they found that HLF overexpression was in fact associated with hypomethylation of its promoter, a feature encountered more commonly in AMLs with DNMT3A R882 mutations than in those with wild-type DNMT3A.
Having identified HLF overexpression to be characteristic of triple-mutant AML, Garg et al investigated whether this had a pathogenetic role by using xenotransplantation into immunodeficient mice. They report that genetic disruption of HLF with CRISPR-single guide RNA (sgHLF) in a human triple-mutant AML xenograft was associated with reduction in the LSC-rich GPR56+CD34+ and expansion in the LSC-poor GPR56–CD34– cell compartments. This phenotype was associated with reduced ability of engrafted sgHLF-targeted AML cells to proliferate when reintroduced into culture ex vivo and a further reduction in the GPR56+CD34+ compartment upon secondary transplantation.
To understand the mechanistic basis of these leukemogenic effects of HLF overexpression, the authors used label-retention and 5-ethynyl-2-deoxyuridine (EdU) pulse-chase studies to characterize the impact of HLF loss on cell cycle and proliferative properties of AML cells. They report that sgHLF-targeted cells progressed through the cell cycle faster, divided more often, and were more sensitive to anti-AML drugs, including cytarabine and daunorubicin, than were control cells. To delve into the molecular basis for this phenomenon, they studied gene expression changes upon short hairpin RNA–mediated inhibition of HLF in the same triple-mutant AML cells. Among other effects, this resulted in downregulation of the cyclin-dependent kinase inhibitor gene CDKN1C and the NOTCH target gene hairy and enhancer of split 1 (HES1), which are plausible mediators of the ability of HLF overexpression to slow down cell cycle progression and maintain the LSC-rich GPR56+CD34+ compartment, respectively (see figure). Garg et al propose that HLF plays an important leukemogenic role in triple-mutant AML and is likely to be at least in part responsible for the poor prognosis of triple-mutant AML. Thus HLF and its effectors represent possible therapeutic vulnerabilities of this and potentially other AML subtypes.
Beyond providing novel insights into the molecular pathogenesis of AML, the article by Garg et al also represents an important paradigm in the study of carcinogenesis by demonstrating that the combined molecular effects of multiple oncogenic mutations can have unpredictable consequences that can influence cellular phenotype, clinical behavior, and responsiveness to therapy. Researchers and clinicians need to bear this in mind when considering the large number of AML cases driven by other combinations of 3 or more mutations, which are rare or even unique and cannot easily be studied in the same way. Although this is a sobering thought, it may also be safe to assume that the reason some mutations cooccur only rarely is that they are not able to synthesize powerful molecular effects in combination, unlike the 3 mutations studied here.
Conflict-of-interest disclosure: G.S.V. is a consultant to Kymab and OxStem.

This feature is available to Subscribers Only
Sign In or Create an Account Close Modal