

Key Points
Adenosine triggers NETs via engaging specific ARs on neutrophils.
Genetic deficiency of ADA2 results in enhanced adenosine-mediated NET formation and subsequent TNF production in patients with DADA2.

Abstract
Reduction of adenosine deaminase 2 (ADA2) activity due to autosomal-recessive loss-of-function mutations in the ADA2 gene (previously known as CECR1) results in a systemic vasculitis known as deficiency of ADA2 (DADA2). Neutrophils and a subset of neutrophils known as low-density granulocytes (LDGs) have been implicated in the pathogenesis of vasculitis, at least in part, through the formation of neutrophil extracellular traps (NETs). The study objective was to determine whether neutrophils and NETs play a pathogenic role in DADA2. In vivo evidence demonstrated NETs and macrophages in affected gastrointestinal tissue from patients with DADA2. An abundance of circulating LDGs prone to spontaneous NET formation was observed during active disease in DADA2 and were significantly reduced after remission induction by anti–tumor necrosis factor (TNF) therapy. Increased circulating LDGs were identified in unaffected family members with monoallelic ADA2 mutations. Adenosine triggered NET formation, particularly in neutrophils from female patients, by engaging A1 and A3 adenosine receptors (ARs) and through reactive oxygen species– and peptidylarginine deiminase–dependent pathways. Adenosine-induced NET formation was inhibited by recombinant ADA2, A1/A3 AR antagonists, or by an A2A agonist. M1 macrophages incubated with NETs derived from patients with DADA2 released significantly greater amounts of TNF-α. Treatment with an A2AAR agonist decreased nuclear translocation of NF-κB and subsequent production of inflammatory cytokines in DADA2 monocyte-derived macrophages. These results suggest that neutrophils may play a pathogenic role in DADA2. Modulation of adenosine-mediated NET formation may contribute a novel and directed therapeutic approach in the treatment of DADA2 and potentially other inflammatory diseases.
Introduction
Adenosine is a purine nucleoside that plays a complex role in the modulation of inflammation. Extracellular levels of adenosine can rise from nanomolar to micromolar concentrations at sites of inflammation.1-3 Depending on the concentration of adenosine in the microenvironment and the adenosine receptor (AR) profile of the relevant cellular inflammatory infiltrate, adenosine can serve either a pro- or anti-inflammatory function.4 Adenosine signals through 4 widely expressed G-protein–coupled receptors: A1, A2A, A2B, and A3. Experimental studies have shown that selective AR agonists can promote or regulate inflammation, highlighting the need for a precise understanding of the nuanced ways in which adenosine regulates immune responses.
Neutrophils are the most abundant white blood cell in humans and play a crucial role in the innate immune response and acute inflammation. Neutrophils generate and release adenosine at sites of inflammation, and adenosine can either activate or inhibit various neutrophil functions.5 For example, adenosine-mediated stimulation of A1 and A3 ARs on neutrophils promotes chemotaxis and phagocytosis,6-8 whereas stimulation of A2A and A2B receptors dampens neutrophil responses by inhibiting respiratory bust and granule release.9,10
Neutrophil extracellular trap (NET) formation is a form of cell death characterized by the extracellular release of granule proteins bound to a decondensed chromatin meshwork.11 NETs can trap and facilitate the elimination of various pathogens, highlighting their putative role in host defense. Although NET formation can serve a protective function to the host, dysregulation in this pathway can cause tissue damage and inflammation.12 In systemic lupus erythematosus (SLE) and antineutrophil cytoplasmic antibody (ANCA)-associated vasculitis, enhanced NET formation can promote vascular disease and inflammation.13-16 Various sterile and nonsterile stimuli can trigger NET formation; however, it is currently unknown whether adenosine can stimulate neutrophils to release NETs.
Deficiency of adenosine deaminase 2 (DADA2) is a recessively inherited autoinflammatory disorder due to loss-of-function mutations in the ADA2 gene.17,18 The ADA2 gene encodes a dimeric protein that is secreted into the extracellular space where it may function as a deaminase to reduce adenosine levels at the site of inflammation.19 In support of this hypothesis, high levels of adenosine deaminase 2 (ADA2) are found in plasma samples and pleural effusions of patients with infectious diseases or chronic inflammation.20-23 Intracellular adenosine is primarily deaminated by ADA1. In addition, ADA2 shares high sequence similarity with adenosine deaminase growth factors (ADGFs) and thus has a role in cell differentiation independent of its catalytic activity.24 Features of DADA2 include vasculitis of predominantly small- to medium-sized blood vessels and other forms of vasculopathy, including livedo reticularis and stroke.25 An abundance of neutrophils and macrophages has been observed at sites of inflammation in biopsies from patients with DADA2.26 DADA2 can skew differentiation of monocytes toward proinflammatory M1 macrophages, but the effects of ADA2 deficiency on neutrophil differentiation and function have not been investigated.17 Because DADA2 is a disease caused by deficiency of an enzyme that may regulate the extracellular concentration of adenosine, the objective of this study was to examine the effects of adenosine on neutrophil-mediated inflammation in patients with DADA2 and their family members. We hypothesized that adenosine can trigger NET formation via specific subcellular events, and that enhanced NET formation in blood and tissue contributes to the pathogenesis of DADA2, at least in part, by promoting inflammatory macrophage responses.
Materials and methods
Human subjects
Patients, aged 5 years or older, with DADA2 confirmed by genetic testing and unaffected family members who were heterozygous for mutations in the ADA2 gene were recruited into an observational cohort at the National Institutes of Health (NIH; Bethesda, MD). All clinical assessments were performed by the investigative team on the same day as sample collection. Additional tissue from clinically indicated biopsies was used for research purposes. All patients provided written informed consent, and a local ethics board approved the research (NCT02257866). Blood from healthy volunteers was obtained via the NIH program for healthy volunteers.
Neutrophil and NET isolation
Neutrophils were isolated as previously described.27 Low-density granulocytes (LDGs) were isolated from the peripheral blood mononuclear cell (PBMC) layer using negative selection.13 Neutrophils and LDGs were resuspended in RPMI and seeded in 24-well plates to generate spontaneous NETs. After 3 to 4 hours, NETs were digested with 10 U/mL micrococcal nuclease (Thermo, Waltham, MD) for 15 minutes at 37°C. NETs were collected and cleared of debris by centrifugation.
Immunofluorescence
Neutrophils were fixed in 4% paraformaldehyde (PFA) in phosphate-buffered saline (PBS) overnight at 4°C, washed, and blocked with 0.2% porcine gelatin (Sigma, St. Louise, MO) for 30 minutes, then incubated with primary antibody for 1 hour in a humid chamber at 37°C. Coverslips were then washed 3 times and incubated 30 minutes with secondary antibody at 37°C. Nuclei were counterstained with 1:1000 Hoechst at room temperature. After washing 3 more times, coverslips were mounted on glass slides using Prolong-gold solution (Invitrogen). Images were acquired on a Zeiss LSM 780 confocal microscope.
Detection of NETs in tissue
Paraffin-embedded slides were deparaffinized with xylene for 20 minutes at room temperature. Tissue was dehydrated through a gradient of ethanol (100%, 95%, 70%, 50%), then rehydrated in water followed by antigen retrieval for 3 minutes. Tissue was blocked with 10% bovine serum albumin (BSA) for 1 hour, then incubated with primary antibody diluted in 5% BSA overnight at 4°C. Samples were washed 3 times with PBS and incubated with secondary antibody at a concentration of 1:400 for 1 hour at room temperature. Nuclei were counterstained with Hoechst (1:1000) for 10 minutes and slides were washed 3 more times with PBS. Images were acquired on a Zeiss LSM 780 confocal microscope.
Statistical analysis
All analyses were performed using GraphPad Prism version 7.0c (La Jolla, CA). The Mann-Whitney U test was used as applicable. All analyses were considered statistically significant at P < .05.
Results
NETs are present in affected tissue and in circulation in patients with DADA2
Histological analysis of surgical resection specimens from the small bowel of a patient with DADA2 revealed myeloid-predominant inflammation in areas of mesenteric arteritis (Figure 1A-B). Prominent NET formation, assessed by citrullinated histone 4 expression, was observed at sites of neutrophilic inflammation in the affected small bowel (Figure 1C). We evaluated the presence of LDGs that are found highly increased in other autoimmune/autoinflammatory conditions, including some forms of vasculitis.28 LDGs are neutrophils that colocalize with PBMCs on density-gradient preparations and in ex vivo experiments display an enhanced capacity to form NETs in the absence of added stimulation.13,14 In patients with DADA2, circulating LDGs were increased during periods of clinically active disease and significantly decreased during clinical remission (Figure 1D). Baseline clinical features of the cohort are detailed in supplemental Table 1 (available on the Blood Web site). Most patients had clinical features that resembled polyarteritis nodosa, including skin involvement (92%) and medium-vessel vasculitis (77%), rather than profound hematologic manifestations or primary immunodeficiency.

NETs are present in DADA2-affected tissue and adenosine induces NET formation through NOX- and PAD-dependent pathways. (A) Hematoxylin-and-eosin (H&E) staining of small bowel tissue obtained from a patient with DADA2 shows neutrophilic infiltration in the wall of mesenteric arteries. (B) Neutrophil (arrows) infiltration in the appendix of a patient with DADA2. Red represents neutrophil elastase and blue is Hoechst. Scale bar, 100 µm. (C) Detection of NETs in a biopsy from a patient with DADA2. Red represents citrullinated histone H4 and blue is Hoechst. Scale bar, 100 µm. (D) LDGs were identified in patients with DADA2. LDGs per milliliter were significantly more abundant in patients with DADA2 during periods of disease activity (active, n = 4; remission, n = 10). LDGs from control (Ctrl; n = 2) and patients with SLE (n = 9) were used for comparison. (E) Adenosine levels were measured in plasma from patients with DADA2 (n = 9) and controls (n = 4). (F-G) Control neutrophils were incubated with different concentrations of adenosine (Ado) for 3 to 4 hours. Immunofluorescence shows that adenosine induces NET formation in control neutrophils, blue represent Hoechst; red is myeloperoxidase (MPO). Scale bar, 50 µm. Results are expressed as percentage of NETs (number of NETs/total number of neutrophils + NETs). **P < .01, Mann-Whitney U test. Results are the means ± standard error of the mean (SEM) of 4 independent experiments. (H) Control neutrophils were incubated in the presence or absence of NOX inhibitor (DPI; 5µM) or pan-PAD inhibitor (Cl-amidine, Cl-am; 20 µM) and stimulated with 16 µM adenosine (Ado). Phorbol myristate acid (PMA; 100 ng/mL) and calcium ionophore (Io; 2.5 µM) were used as positive controls. Results are expressed as percentage of NETs (number of NETs/total number of neutrophils + NETs). **P < .01, ***P < .001, Mann-Whitney U test. Results are the means ± SEM of n = 6. DMSO, dimethyl sulfoxide.
NETs are present in DADA2-affected tissue and adenosine induces NET formation through NOX- and PAD-dependent pathways. (A) Hematoxylin-and-eosin (H&E) staining of small bowel tissue obtained from a patient with DADA2 shows neutrophilic infiltration in the wall of mesenteric arteries. (B) Neutrophil (arrows) infiltration in the appendix of a patient with DADA2. Red represents neutrophil elastase and blue is Hoechst. Scale bar, 100 µm. (C) Detection of NETs in a biopsy from a patient with DADA2. Red represents citrullinated histone H4 and blue is Hoechst. Scale bar, 100 µm. (D) LDGs were identified in patients with DADA2. LDGs per milliliter were significantly more abundant in patients with DADA2 during periods of disease activity (active, n = 4; remission, n = 10). LDGs from control (Ctrl; n = 2) and patients with SLE (n = 9) were used for comparison. (E) Adenosine levels were measured in plasma from patients with DADA2 (n = 9) and controls (n = 4). (F-G) Control neutrophils were incubated with different concentrations of adenosine (Ado) for 3 to 4 hours. Immunofluorescence shows that adenosine induces NET formation in control neutrophils, blue represent Hoechst; red is myeloperoxidase (MPO). Scale bar, 50 µm. Results are expressed as percentage of NETs (number of NETs/total number of neutrophils + NETs). **P < .01, Mann-Whitney U test. Results are the means ± standard error of the mean (SEM) of 4 independent experiments. (H) Control neutrophils were incubated in the presence or absence of NOX inhibitor (DPI; 5µM) or pan-PAD inhibitor (Cl-amidine, Cl-am; 20 µM) and stimulated with 16 µM adenosine (Ado). Phorbol myristate acid (PMA; 100 ng/mL) and calcium ionophore (Io; 2.5 µM) were used as positive controls. Results are expressed as percentage of NETs (number of NETs/total number of neutrophils + NETs). **P < .01, ***P < .001, Mann-Whitney U test. Results are the means ± SEM of n = 6. DMSO, dimethyl sulfoxide.
Adenosine induces NET formation
DADA2-associated mutations lead to absent or decreased activity of ADA2, a secreted enzyme that may degrade extracellular adenosine.17 Adenosine levels were significantly elevated in DADA2 patients when compared with control samples with concentrations ranging from 0.2 to 0.6 µM (Figure 1E). We therefore hypothesized that enhanced NET formation in DADA2 could be mediated by adenosine. To assess the effect of adenosine on NET formation, healthy donor peripheral blood neutrophils were incubated with graded concentrations of adenosine (range, 0.2-16 µM) for 4 hours. Immunofluorescence analysis showed that adenosine significantly increased NET formation compared with untreated neutrophils across the range of all studied concentrations (Figure 1F-G). The NADPH oxidase (NOX) pathway and peptidylarginine deiminase-4 (PAD4) have been implicated in the formation of NETs following various, but not all, stimuli.29-31 To address whether adenosine requires functional NOX and/or PADs to induce NET formation, cells were pretreated with diphenylene iodonium (DPI; NOX inhibitor) at doses that inhibit NOX but not mitochondrial reactive oxygen species (ROS), or with Cl-amidine, a pan-PAD inhibitor that targets PAD4. Confocal analysis showed significant decreases in NET formation after inhibition of NOX or PADs following adenosine stimulation (Figure 1H), suggesting that adenosine induces NETs through NOX- and PAD-dependent pathways. Taken together, these results provide in vivo evidence of enhanced NETosis in patients with DADA2 mediated by extracellular adenosine.
Reduced activity of ADA2 leads to NET formation
Mechanisms leading to the generation of adenosine are pleiotropic, yet specific enzymes help maintain extracellular and intracellular adenosine levels in equilibrium. We hypothesized that accumulation of extracellular adenosine due to genetic deficiency of ADA2 may promote adenosine-mediated NET formation in patients with DADA2. To test that hypothesis, healthy volunteer neutrophils were incubated with adenosine in the presence or absence of recombinant ADA2. ADA2 significantly decreased NET generation induced by adenosine (Figure 2A). Immunofluorescence analyses confirmed the inhibitory effect of recombinant ADA2 on the adenosine effect on neutrophils (Figure 2B). Monocytes and macrophages are the primary source of extracellular ADA2, whereas neutrophils do not synthesize ADA2.32 Thus, we hypothesized that interactions between macrophages and neutrophils are necessary to regulate extracellular concentrations of adenosine. To prove this concept, supernatants were collected from monocyte-derived macrophages generated from healthy volunteers or from DADA2 patients. ADA2 enzyme activity was significantly decreased in supernatants from DADA2 macrophages when compared with supernatant from control macrophages (Figure 2C). Control neutrophils were incubated in the presence of healthy control or DADA2 macrophage supernatants followed by adenosine stimulation. Adenosine-induced NETs were not detected in neutrophils incubated with healthy control macrophage supernatants. In contrast, enhanced NET formation was observed in adenosine-stimulated neutrophils incubated in the presence of supernatant from DADA2 macrophages (Figure 2D-E), suggesting that ADA2 released from macrophages metabolized exogenous adenosine and decreased adenosine-induced NET formation. To corroborate whether this effect was specific to ADA2, recombinant ADA1 or ADA2 was added to DADA2 neutrophils in the presence of exogenous adenosine. NET formation was decreased in neutrophils incubated with human recombinant ADA2 but not in those incubated with ADA1 (Figure 2F). Activity of ADA1 was assessed to corroborate that the enzyme was active (Figure 2G). These results demonstrate that interactions between macrophages and neutrophils are critical to regulate adenosine-mediated NET formation and highlight a potential mechanism whereby deficiency of ADA2 production by macrophages may contribute to dysregulation of adenosine-mediated NET formation in DADA2.

ADA2 decreases adenosine-induced NETs. Control neutrophils were incubated with different concentrations of adenosine (Ado) and in the presence or absence of 100 ng of recombinant ADA2 for 3 to 4 hours. (A) Percentage of NETs were graphed (number of NETs/total number of neutrophils + NETs). Results are the means ± SEM of n = 4, *P < .05, Mann-Whitney U test. (B) Representative confocal images after treatment with recombinant ADA2. Blue represents Hoechst; red is myeloperoxidase (MPO). Control neutrophils were incubated in the presence or absence of supernatant from control macrophages or DADA2 macrophages in the presence of adenosine. Scale bar, 50 µm. (C) ADA2 activity was measured in supernatants from control and DADA2 macrophages after 48-hour incubation. Results are the means ± SEM of n = 4, *P < .05, Mann-Whitney U test. (D) Representative confocal image of neutrophils treated with adenosine in the presence of macrophage supernatant from healthy volunteers or patients with DADA2. Scale bar, 50 µm. (E) Percentage of NETs were graphed (number of NETs/total number of neutrophils + NETs). Results are the means ± SEM of n = 3 to 4, *P < .05, Mann-Whitney U test. (F) Confocal images of DADA2 neutrophils treated with adenosine (Ado) in the presence or absence of human 100 ng of recombinant ADA1 (hrADA1) or ADA2 (hrADA2) for 3 to 4 hours. Blue represents Hoechst; red is myeloperoxidase (MPO). Scale bar, 50 µm. (G) ADA1 activity was measured for the human recombinant ADA1 used. N.D., not determined.
ADA2 decreases adenosine-induced NETs. Control neutrophils were incubated with different concentrations of adenosine (Ado) and in the presence or absence of 100 ng of recombinant ADA2 for 3 to 4 hours. (A) Percentage of NETs were graphed (number of NETs/total number of neutrophils + NETs). Results are the means ± SEM of n = 4, *P < .05, Mann-Whitney U test. (B) Representative confocal images after treatment with recombinant ADA2. Blue represents Hoechst; red is myeloperoxidase (MPO). Control neutrophils were incubated in the presence or absence of supernatant from control macrophages or DADA2 macrophages in the presence of adenosine. Scale bar, 50 µm. (C) ADA2 activity was measured in supernatants from control and DADA2 macrophages after 48-hour incubation. Results are the means ± SEM of n = 4, *P < .05, Mann-Whitney U test. (D) Representative confocal image of neutrophils treated with adenosine in the presence of macrophage supernatant from healthy volunteers or patients with DADA2. Scale bar, 50 µm. (E) Percentage of NETs were graphed (number of NETs/total number of neutrophils + NETs). Results are the means ± SEM of n = 3 to 4, *P < .05, Mann-Whitney U test. (F) Confocal images of DADA2 neutrophils treated with adenosine (Ado) in the presence or absence of human 100 ng of recombinant ADA1 (hrADA1) or ADA2 (hrADA2) for 3 to 4 hours. Blue represents Hoechst; red is myeloperoxidase (MPO). Scale bar, 50 µm. (G) ADA1 activity was measured for the human recombinant ADA1 used. N.D., not determined.
A1 and A3 ARs mediate adenosine-induced NET formation
Adenosine exerts its biologic function by engaging a family of 4 cell surface P1 purinergic receptors (A1, A2A, A2B, and A3).33 A1AR and A3AR are coupled to the Gi protein α-subunit and inhibit the formation of cellular cAMP. A2AAR and A2BAR are Gs coupled and stimulate adenylyl cyclase with resultant increased production of cAMP-dependent proteins. Because adenosine can engage multiple ARs, we used pharmacologic approaches to determine the receptor subtype(s) responsible for adenosine-mediated NET formation. Healthy control neutrophils were preincubated with various antagonists of A1AR (5 nM), A2AAR (5 nM), or A3AR (5 nM) prior to adenosine stimulation. Immunofluorescence analysis showed that NET formation induced by adenosine was significantly abrogated in the presence of A1AR and A3AR antagonists but not an A2AAR antagonist (Figure 3A-B). These results suggest that A1AR and A3AR mediate adenosine-induced NET formation. To corroborate that A1AR and A3AR are involved in adenosine-induced NET formation, control neutrophils were incubated with specific A1AR (5 nM 5-Deoxy-(±)-ENBA [Cl-ENBA](N-bicyclo[2.2.1]hept-2-yl-5'-chloro-5'-deoxyadenosine)) and A3AR (100 nM; MRS5698) agonists. The A1AR, but not the A3AR, agonist significantly induced NET formation (Figure 3C). To corroborate the role of A1AR in triggering NET formation, different concentrations of A1AR agonist were used. NETs were triggered by the A1AR agonist even at concentrations as low as 5 pM (Figure 3D). To further prove the involvement of A1AR in NET formation, neutrophils were incubated with Cl-ENBA in the presence or absence of highly specific A1AR antagonists, DPCPX (8-cyclopentyl-1,3-dipropylxanthine) and WRC0571. DPCPX decreased Cl-ENBA-induced NET formation in a concentration-dependent manner reaching a full inhibition at nanomolar concentrations (Figure 3E). WRC0571 was equally effective and diminished the rate of NETting cells down to background level (Figure 3E). These results suggest that the A1AR is the dominant receptor in triggering NET formation by adenosine.
![Figure 3. A1 and A3 ARs mediate adenosine-induced NETosis. Control neutrophils were incubated with adenosine (Ado) in the presence or absence of different AR antagonist for 3 to 4 hours. (A) Percentage of NETs was quantified (number of NETs/total number of neutrophils + NETs). Results are representative of 4 different experiments. (B) Representative confocal images of NET formation after treatment with an AR antagonist. Blue represents Hoechst; red is myeloperoxidase (MPO). Scale bar, 50 µm. (C) Neutrophils were incubated with adenosine, A1AR (5-deoxy-(±)-ENBA [Cl-ENBA]) or A3AR (MRS5698) agonists for 2 hours. NETs were quantified. (D-E) NETs were quantified after different concentrations of an A1AR agonist and/or in the presence of specific A1AR antagonists. *P < .05, **P < .01, ***P < .001, ****P < .0001, Mann-Whitney U test. Results are the means ± SEM of n = 4.](https://ash.silverchair-cdn.com/ash/content_public/journal/blood/134/4/10.1182_blood.2018892752/3/m_blood892752f3.png?Expires=1769083072&Signature=XQY52TDUsXZwtEKqFJZ462garDGL2q7ppcIwGtEBsjBt-ZiZ-TxtuLMkV3woevK4MEqXfWbkY54iFpcl0G3JD1BNUHnDw3YLhNPN-NGZMA~GYHa4AbFQnsD9gXaDWAJwcotf0T5-dgNfHIZ9zjmQrKYYM6P3JbuzjYz3x0mCJAUsJDf8CPZOKUd-hC6q7MywexCJXKwPeyOShwwS5ZjCBoEdP~gyp4yvYpZ2KjmJ-OvEXoHgWxSrPtIbsZi4Hafl1IHpB0McOp5J6uN~g~aPO6z2ZMSMZCm1E53YskCrR43eRg0~OcFJRa7Vgr4U61H3qNShLS5WQ2V5fWOquhmp5g__&Key-Pair-Id=APKAIE5G5CRDK6RD3PGA)
A1 and A3 ARs mediate adenosine-induced NETosis. Control neutrophils were incubated with adenosine (Ado) in the presence or absence of different AR antagonist for 3 to 4 hours. (A) Percentage of NETs was quantified (number of NETs/total number of neutrophils + NETs). Results are representative of 4 different experiments. (B) Representative confocal images of NET formation after treatment with an AR antagonist. Blue represents Hoechst; red is myeloperoxidase (MPO). Scale bar, 50 µm. (C) Neutrophils were incubated with adenosine, A1AR (5-deoxy-(±)-ENBA [Cl-ENBA]) or A3AR (MRS5698) agonists for 2 hours. NETs were quantified. (D-E) NETs were quantified after different concentrations of an A1AR agonist and/or in the presence of specific A1AR antagonists. *P < .05, **P < .01, ***P < .001, ****P < .0001, Mann-Whitney U test. Results are the means ± SEM of n = 4.
A1 and A3 ARs mediate adenosine-induced NETosis. Control neutrophils were incubated with adenosine (Ado) in the presence or absence of different AR antagonist for 3 to 4 hours. (A) Percentage of NETs was quantified (number of NETs/total number of neutrophils + NETs). Results are representative of 4 different experiments. (B) Representative confocal images of NET formation after treatment with an AR antagonist. Blue represents Hoechst; red is myeloperoxidase (MPO). Scale bar, 50 µm. (C) Neutrophils were incubated with adenosine, A1AR (5-deoxy-(±)-ENBA [Cl-ENBA]) or A3AR (MRS5698) agonists for 2 hours. NETs were quantified. (D-E) NETs were quantified after different concentrations of an A1AR agonist and/or in the presence of specific A1AR antagonists. *P < .05, **P < .01, ***P < .001, ****P < .0001, Mann-Whitney U test. Results are the means ± SEM of n = 4.
Neutrophil AR expression differs by sex in patients with DADA2
We observed that adenosine-mediated NETosis was more robust in neutrophils from female patients with DADA2 (supplemental Figure 1A-B). Because adenosine primarily engages A1AR, and to a lesser extent A3AR, in neutrophils to form NETs, we quantified the adenosine subtype receptor density on the surface of neutrophils from patients with DADA2 stratified by sex. Flow analysis showed 3 different neutrophil populations for A1AR, classified as negative, low-, and high-expressing neutrophils (Figure 4A-B). Female patients with DADA2 displayed a significantly greater percentage of neutrophils displaying high expression of A1AR receptors compared with male patients with DADA2 (Figure 4C). Male patients had a significantly greater percentage of neutrophils with no A1AR expression compared with neutrophils from female patients (Figure 4C). A greater percentage of A3AR+ cells was observed in female patients compared with male patients (Figure 4D-F). Female healthy control neutrophils also displayed significantly greater percentages of neutrophils positive for A1AR and A3AR when compared with healthy male neutrophils (Figure 4G-H). These data underlie sex differences of AR expression on neutrophils and may explain why neutrophils from female patients undergo more robust NET formation in response to adenosine.

Sex differences in AR expression on neutrophils. ARs were detected in neutrophils from patients with DADA2. (A,D) Confocal analysis shows the presence of A1AR and A3AR (in red) in the plasma membrane of DADA2 neutrophils. Nuclei were stained in blue. Scale bar, 5 µm. (B) A1AR was analyzed in neutrophils from patients with DADA2 by flow cytometry. Three populations of A1AR were found in neutrophils from patients with DADA2. (C) Percentage of neutrophils with surface A1AR, classified as negative, low, and high. (E) A3AR was analyzed in neutrophils from patients with DADA2 by flow cytometry. (F) Percentage of neutrophil for A3AR. (G) A1AR and (H) A3AR were analyzed in neutrophils from healthy with by flow cytometry. *P < .05 Mann-Whitney U test.
Sex differences in AR expression on neutrophils. ARs were detected in neutrophils from patients with DADA2. (A,D) Confocal analysis shows the presence of A1AR and A3AR (in red) in the plasma membrane of DADA2 neutrophils. Nuclei were stained in blue. Scale bar, 5 µm. (B) A1AR was analyzed in neutrophils from patients with DADA2 by flow cytometry. Three populations of A1AR were found in neutrophils from patients with DADA2. (C) Percentage of neutrophils with surface A1AR, classified as negative, low, and high. (E) A3AR was analyzed in neutrophils from patients with DADA2 by flow cytometry. (F) Percentage of neutrophil for A3AR. (G) A1AR and (H) A3AR were analyzed in neutrophils from healthy with by flow cytometry. *P < .05 Mann-Whitney U test.
NETs induce NF-κB nuclear translocation and TNF-α release in macrophages
Polarization of monocytes into M1 macrophages with subsequent production of tumor necrosis factor (TNF) has been implicated in the pathogenesis of DADA2.17 However, the specific factors mediating enhanced proinflammatory cytokine production in DADA2 macrophages remain unclear. TNF-α+ cells and tissue deposition of this cytokine were evident in DADA2 (Figure 5A). To test whether NETs can directly shape macrophage effector responses in DADA2, control M1 macrophages were incubated with spontaneously generated NETs derived from DADA2 LDGs or normal density neutrophils. After 45 minutes to 2 hours of incubation, the NF-κB p65 subunit translocated into the nucleus of those macrophages exposed to DADA2 NETs (Figure 5B-C), followed by enhanced TNF-α release after 48 hours (Figure 5D). To corroborate in vivo the potential interaction between macrophages and NETs, affected appendix tissue from a patient with DADA2 was examined. Confocal microscopy analysis demonstrated spatial proximity of NETs and macrophages in the affected tissue (Figure 5E), suggesting that NETs can stimulate macrophages to release TNF-α in vivo in patients with DADA2.

NETs promote NF-κB nuclear translocation and stimulation of TNF-α release from macrophages. (A) Neutrophils and TNF-α were detected in tissue from a patient with DADA2. Red represents elastase; green is TNF-α; and blue is Hoechst. Scale bar, 100 µm. Control macrophages were incubated in presence of spontaneously generated NETs from LDGs or normal dense neutrophils from DADA2 patients or lipopolysaccharide (LPS) for 45 minutes to 2 hours. (B-C) Translocation of the NF-κB subunit p65 was assessed by immunofluorescence and quantified. LPS was used as positive control. Scale bar, 10 µm. (D) Levels of TNF-α in supernatants from control and DADA2 macrophages incubated with NETs for 48 hours were analyzed by enzyme-linked immunosorbent assay (ELISA). *P < .05, **P < .01, ****P < .0001, Mann-Whitney U test. Results are the means ± SEM of 6 independent experiments. (E) Macrophages and NETs detected in appendix tissue from a patient with DADA2. Red represents citrullinated histone H4; green is a marker for macrophages; and blue is Hoechst. Scale bar, 10 µm.
NETs promote NF-κB nuclear translocation and stimulation of TNF-α release from macrophages. (A) Neutrophils and TNF-α were detected in tissue from a patient with DADA2. Red represents elastase; green is TNF-α; and blue is Hoechst. Scale bar, 100 µm. Control macrophages were incubated in presence of spontaneously generated NETs from LDGs or normal dense neutrophils from DADA2 patients or lipopolysaccharide (LPS) for 45 minutes to 2 hours. (B-C) Translocation of the NF-κB subunit p65 was assessed by immunofluorescence and quantified. LPS was used as positive control. Scale bar, 10 µm. (D) Levels of TNF-α in supernatants from control and DADA2 macrophages incubated with NETs for 48 hours were analyzed by enzyme-linked immunosorbent assay (ELISA). *P < .05, **P < .01, ****P < .0001, Mann-Whitney U test. Results are the means ± SEM of 6 independent experiments. (E) Macrophages and NETs detected in appendix tissue from a patient with DADA2. Red represents citrullinated histone H4; green is a marker for macrophages; and blue is Hoechst. Scale bar, 10 µm.
A2AAR agonist decreases nuclear NF-κB translocation and NET-induced proinflammatory cytokines in macrophages
Because DADA2 NETs can induce NF-κB activation and TNF-α release in macrophages, we tested whether modulation of adenosine-signaling pathways reduces macrophage activation by NETs. Because activation of A2AAR can promote anti-inflammatory responses in myeloid cells, M1 macrophages were pretreated with an A2AAR agonist prior to stimulation with NETs. After preincubation with an A2AAR agonist for 30 minutes, macrophages were incubated with spontaneously generated NETs from DADA2 LDGs or normal dense neutrophils. A2AAR agonist decreased both NET-induced nuclear NF-κB translocation and proinflammatory cytokine gene expression, including TNFA, IL6, and IL8, in macrophages (Figure 6). These results support the concept that modulation of interactions between NETs and macrophages or specific targeting of adenosine pathways could have therapeutic efficacy in DADA2.
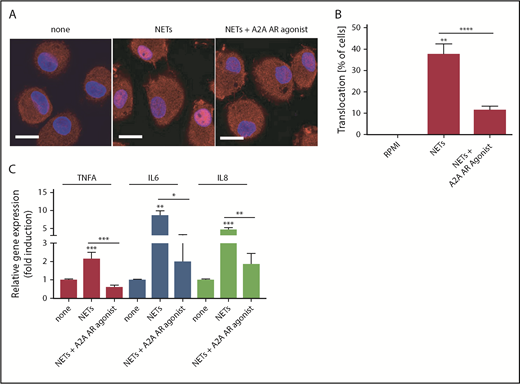
An A2AAR agonist decreases NF-κB nuclear translocation and NET-induced proinflammatory cytokine production in macrophages. Control macrophages were incubated with NETs in the presence or absence of A2AAR agonist. (A) Translocation of NF-κB (in red) was impaired in cells treated A2AAR agonist. Nuclei were stained in blue. Scale bar, 10 µm. (B) Translocation of NF-κB was quantified and graphed as a percentage of total cells. (C) Gene expression analysis shows A2AAR agonist significantly decreased NET-induced proinflammatory cytokine in macrophages. *P < .05, **P < .01, ***P < .001, ****P < .0001, Mann-Whitney U test. Results are the means ± SEM of n = 4-5.
An A2AAR agonist decreases NF-κB nuclear translocation and NET-induced proinflammatory cytokine production in macrophages. Control macrophages were incubated with NETs in the presence or absence of A2AAR agonist. (A) Translocation of NF-κB (in red) was impaired in cells treated A2AAR agonist. Nuclei were stained in blue. Scale bar, 10 µm. (B) Translocation of NF-κB was quantified and graphed as a percentage of total cells. (C) Gene expression analysis shows A2AAR agonist significantly decreased NET-induced proinflammatory cytokine in macrophages. *P < .05, **P < .01, ***P < .001, ****P < .0001, Mann-Whitney U test. Results are the means ± SEM of n = 4-5.
Neutrophils from DADA2 first-degree relatives display dysregulation in NET formation
Family members with monoallelic pathogenic mutations can have reduced ADA2 functional activity, albeit to a lesser degree than DADA2 patients with biallelic mutations.17 Additionally, autoimmunity has been reported in some of the family members who are carriers of ADA2 mutations.34 In this cohort, a maternal great-grandmother (ADA2 mutation carrier) of 2 children with genetically confirmed DADA2 (compound heterozygous mutations) was diagnosed with adult-onset, biopsy-proven polyarteritis nodosa, a disease that shares strong clinical resemblance to DADA2 (supplemental Figure 2A). We tested the hypothesis that family members of patients with DADA2 may have neutrophil function abnormalities in the absence of overt disease features. First-degree relatives of patients with DADA2, who were studied during a period of apparent normal health, displayed significantly increased circulating LDGs compared with controls (supplemental Figure 2B). In addition, neutrophils isolated from 2 families showed enhanced NET formation in female family members with monoallelic mutations in the ADA2 gene compared with males, consistent with the previous observation in DADA2 patients that the effect of adenosine in NET formation may be determined in part by sex (supplemental Figure 2C).
Discussion
The discovery of monogenic inflammatory diseases creates unique opportunities to understand how genetic mutations and perturbations in specific metabolic pathways can lead to inflammation in human disease. The role of adenosine in the regulation inflammation in humans is complex, as adenosine is challenging to study in vitro and can serve both pro- and anti-inflammatory functions. DADA2, a multisystem disease defined by loss-of-function mutations in ADA2, an enzyme that metabolizes extracellular adenosine, offers an opportunity to learn how specific defects of adenosine metabolism can lead to enhanced inflammation. In the absence of ADA2, patients with DADA2 develop a complex clinical phenotype that includes a polyarteritis nodosa–like vasculitis that targets the skin and gastrointestinal tract, early-onset stroke, and a range of other manifestations including hematologic abnormalities.35-37 Our findings confirm that neutrophils likely play a role in DADA2 pathogenesis, adding this monogenic form of vasculitis to an expanding list of systemic inflammatory diseases characterized by enhanced NET formation.38
In the present study, we explore the potential role of neutrophils in DADA2 and report for the first time that NET formation is enhanced in DADA2 and is related to the effect of adenosine on neutrophils in the absence of ADA2. These findings add to the understanding of the pathophysiology of DADA2, the response of neutrophils to adenosine, and the role of ADA2 in mediating interactions between macrophages and neutrophils. Alterations in adenosine metabolism likely contribute to neutrophil-mediated inflammation in DADA2. We demonstrated enhanced NET formation in patients with DADA2 in circulation, in the form of LDGs, and in affected tissue. Because patients under anti-TNF treatment displayed significantly reduced LDG numbers, it is possible that TNF-α or other proinflammatory cytokines can contribute to the presence of LDGs but further studies are needed to address these questions. In vitro experiments suggest that enhanced NET formation in DADA2 is a consequence of dysregulated adenosine metabolism, higher expression of A1 and A3 receptors on the surface of neutrophils, and activation of NET canonical pathways.
Adenosine can regulate neutrophil function.5,39 Submicromolar concentrations of adenosine can stimulate high-affinity A1 and A3 receptors to promote neutrophil chemotaxis toward inflammatory stimuli. Activation of A2A and lower-affinity A2B receptors on neutrophils with increasing concentrations of adenosine can inhibit neutrophil adhesion and migration across the endothelial cell barrier. These findings have been used to promote the hypothesis that increasing concentrations of adenosine may shift the neutrophil away from proinflammatory and toward anti-inflammatory responses; however, this concept is likely an oversimplification. For example, activation of both low- and high-affinity ARs can inhibit release of proinflammatory cytokines from activated neutrophils (A2A and A1 receptors)6,40 or inhibit degranulation (A2A and A3 receptors).10,41-43 Challenges inherent to the study of neutrophils (eg, short-lived cells prone to cell death that may variably increase extracellular adenosine levels during in vitro experiments) and lack of specificity of currently available adenosine subtype receptor agonists/antagonists complicate these studies. In the present study, we report that adenosine can trigger NET formation, and this is inhibited by antagonism of A1 or A3 receptors. These findings support existing evidence that activation of A1 and A3 receptors can promote proinflammatory neutrophil responses and extend understanding of how adenosine can modulate neutrophil function to include NET formation, but do not support the concept that adenosine-mediated NETosis via activation of A1 or A3 receptors occurs exclusively at lower concentrations of extracellular adenosine.
We identified sex-specific differences of adenosine subtype receptors on both DADA2 and healthy control neutrophils. There are few reports on sex differences in clinical response to adenosine and distribution of ARs. Previous studies examining the safety and tolerability of IV adenosine infusion for cardiac stress-imaging studies have demonstrated an increased physiologic response and side effects in female patients.44,45 Increased A1 receptors have been observed in female vs male cardiac tissue in C57BL/6 mice.46 Depletion of A1 or A2B receptors protects against salt-induced hypertension in female but not male rats.47 Several rheumatologic diseases have strong female predilection and are associated with enhanced NET formation, including SLE and rheumatoid arthritis.48,49 Whether sex-specific differences in the neutrophil-related response to adenosine may play a role in the pathophysiology of these conditions should be explored.
The present study highlights novel aspects of the relationship between neutrophils and macrophages in the pathogenesis of DADA2. Neutrophils are often recruited early to sites of inflammation, with later-stage recruitment of macrophages. Understanding neutrophil-macrophage interactions may therefore provide insight into regulation of chronic inflammation.50,51 Results from the present study demonstrate that macrophage secretion of ADA2 is an important regulator of adenosine-mediated NET formation. Subsequent contact of NETs with macrophages can further activate NF-κB and lead to the production and release of proinflammatory cytokines, including TNF-α. These cytokines may in turn trigger further NET formation thus amplifying the inflammatory response (Figure 7).
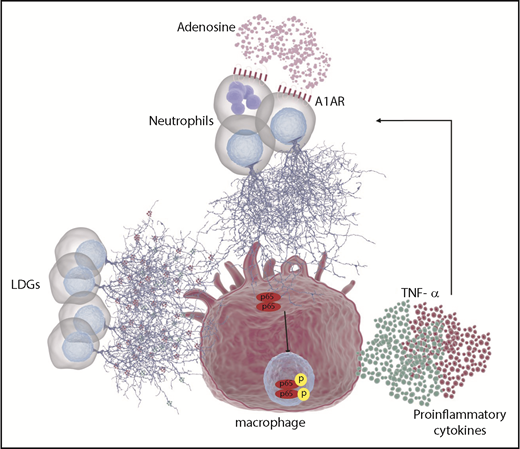
Schematic representation of the role of neutrophil and macrophages in DADA2. Due to the lack of ADA2 activity in patients with DADA2, extracellular adenosine may accumulate. Elevated concentrations of adenosine can engage A1AR and/or A3AR in neutrophils leading to NET formation. In addition, LDGs present in patients with DADA2 release NETs spontaneously. Both sources of NETs can activate macrophages, leading to NF-κB translocation to the nucleus and activation of proinflammatory cytokines such as TNF-α and IL-6, among others. In turn, TNF-α can prime neutrophils to undergo further NETosis, leading to a vicious cycle.
Schematic representation of the role of neutrophil and macrophages in DADA2. Due to the lack of ADA2 activity in patients with DADA2, extracellular adenosine may accumulate. Elevated concentrations of adenosine can engage A1AR and/or A3AR in neutrophils leading to NET formation. In addition, LDGs present in patients with DADA2 release NETs spontaneously. Both sources of NETs can activate macrophages, leading to NF-κB translocation to the nucleus and activation of proinflammatory cytokines such as TNF-α and IL-6, among others. In turn, TNF-α can prime neutrophils to undergo further NETosis, leading to a vicious cycle.
Beyond DADA2, the present study uncovers neutrophil-related abnormalities in family members of patients with this condition who carry monoallelic mutations in the ADA2 gene. An increased abundance of circulating LDGs was observed in healthy parents of patients with DADA2. Reduction of ADA2 enzyme activity has been demonstrated in relatives of DADA2 patients.17 In our cohort, we observed incomplete features of DADA2 and even 1 case of adult-onset biopsy-proven polyarteritis nodosa in association with monoallelic mutations in ADA2. As another example, early-onset thalamic lacunar infarct was discovered incidentally on brain imaging in an otherwise healthy relative of a patient with DADA2. Prospective, observational studies of individuals with heterozygous mutations in the ADA2 gene are needed to determine whether these patients indeed have incomplete forms of DADA2 (eg, increase risk vasculitis and stroke). Similarly, patients with polyarteritis nodosa should be screened for mutations in ADA2, regardless of the age of disease onset, which might be important for guiding their therapy.52
We investigated the function of various ARs with respect to adenosine-mediated NET formation. Due to the lack of specific antibodies for A2bARs, we were only able to study the role of A1ARs, A2AARs, and A3ARs with respect to adenosine-mediated NET formation. Given that A2BARs couple to the same Gs subunit as A2AARs, it is possible that A2BAR stimulation may inhibit NET formation. However, unlike A2AARs, A2BARs are also coupled to the Gq subunit and could thus increase intracellular calcium levels, an important event in the formation of NETs. Generation of a transgenic murine model of DADA2 to study the effects of ADA2 deficiency on neutrophil biology and adenosine-mediated inflammation may help to further understand whether targeting of adenosine-mediated NET formation could be a novel therapeutic approach in DADA2. However, fundamental differences in murine and human neutrophil biology could pose challenges in extrapolating findings into human disease.12
The observations in this study highlight a novel mechanism where adenosine, neutrophils, and macrophages interact to promote a proinflammatory microenvironment in the context of a monogenic form of vasculitis known as DADA2. Targeting specific pathways of adenosine-mediated NET formation or developing strategies that disrupt deleterious neutrophil-macrophage interactions and the generation of NETs may offer new therapeutic approaches to the treatment of DADA2. Modulation of ARs, either through antagonism of A1 or A3 receptors or agonism of the A2A receptor, or supplementation of patients with recombinant pegylated ADA2 could be tested as potential pathway-directed novel therapeutic interventions in DADA2. Finally, targeting molecules in adenosine-mediated signaling pathways and NET formation might be therapeutic for a broad spectrum of human inflammatory diseases.
For data sharing, please e-mail the corresponding authors at carmelo.carmona-rivera@nih.gov or peter.grayson@nih.gov.
The online version of this article contains a data supplement.
The publication costs of this article were defrayed in part by page charge payment. Therefore, and solely to indicate this fact, this article is hereby marked “advertisement” in accordance with 18 USC section 1734.
Acknowledgments
This work was supported by the Intramural Research Program at the National Institutes of Health, National Institute of Arthritis and Musculoskeletal and Skin Diseases (ZIAAR041199) and National Institute of Diabetes and Digestive and Kidney Diseases (ZIADK31117 [K.A.J.]).
Authorship
Contribution: C.C.-R., S.S.K., K.W.S., J.A.I.-C., L.J.O., Y.L., and W.L.T. performed the experiments, analyzed the data, and performed statistical analyses; C.C.-R., S.S.K., K.A.J., D.L.S., A.K.O., D.L.K., I.A., M.J.K., and P.C.G. provided patients, scientific input, and/or manuscript preparation; and C.C.-R. and P.C.G. were involved in overall design and conceptualization.
Conflict-of-interest disclosure: The authors declare no competing financial interests.
Correspondence: Carmelo Carmona-Rivera, Systemic Autoimmunity Branch, National Institute of Arthritis and Musculoskeletal and Skin Diseases, National Institutes of Health, Building 10 Room 12S-253, 10 Center Dr, Bethesda, MD 20892; e-mail: carmelo.carmona-rivera@nih.gov; and Peter C. Grayson, Systemic Autoimmunity Branch, Vasculitis Translational Research Program, National Institute of Arthritis and Musculoskeletal and Skin Diseases, National Institutes of Health, Building 10 Room 12S-253, 10 Center Dr, Bethesda, MD 20892; e-mail: peter.grayson@nih.gov.
