

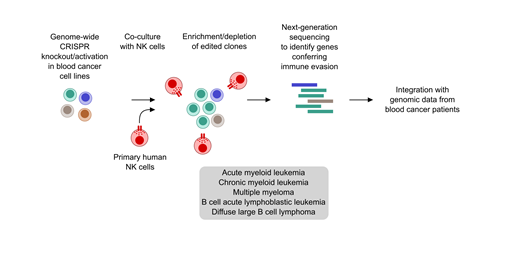
Natural killer (NK) cells have been suggested to control progression and relapse in several hematological malignancies. Enhancing NK cell reactivity represents an attractive approach to improve treatment responses. However, mechanisms enabling evasion of hematologic cancer cells from NK cells are incompletely understood. To identify cancer cell-intrinsic factors enabling resistance to NK cell cytotoxicity, we conducted genome-wide CRISPR screens in a range of hematological malignancies.
Cas9-expressing cancer cells from diverse hematological malignancies, including acute and chronic myeloid leukemia (AML and CML), multiple myeloma (MM), diffuse large B-cell lymphoma (DLBCL), and B cell acute lymphoblastic leukemia were infected with a genome-scale lentiviral sgRNA library and exposed to primary human NK cells. Genes essential for NK cell cytotoxicity were discovered from surviving cancer cells which were enriched with gene knockouts inducing reduced sensitivity to NK cell killing. Additional data from a gain-of-function screen using a genome-scale CRISPR activation system were generated using the MM.1S myeloma cell line. Results from the functional genomic screens were integrated with transcriptomic data from > 9,000 patients across hematological cancers, as well as multi-omics data from AML and DLBCL, and both public and in-house single-cell RNA-sequencing data from AML patients.
Knockout of genes encoding components involved in antigen presentation (B2M, HLA-A, HLA-C, HLA-E, TAP1, TAP2) sensitized multiple blood cancer cell lines to NK cell cytotoxicity, consistent with the inhibitory effect of MHC I on NK cells. Furthermore, knockout of interferon and JAK-STAT signaling components sensitized cancer cells to NK cell lysis, suggesting that MHC class I induction in response to interferon promotes NK cell resistance across cancer types.
Interestingly, genes and pathways whose disruption conferred resistance for NK cell-mediated lysis exhibited partial overlap but also substantial variability across blood cancer types. Knockout of NCR3LG1 (B7-H6, ligand for the NKp30 activating NK cell receptor), resulted in resistance of K562 CML cells to NK cell cytotoxicity. In contrast, disruption of genes encoding apoptotic mediators (FADD, PMAIP1, BID) and TRAIL receptors (TNFRSF10B) conferred resistance to NK cell cytotoxicity in SUDHL4 DLBCL cells. The same pathways were identified in the MM cell line MM.1S, in which knockout of FAS, CASP2, and CASP8 as well as the TRAIL receptor TNFRSF10A induced NK cell resistance. Furthermore, loss of CD48, a ligand of the non-MHC binding receptor CD244 on NK cell surface, resulted in resistance and a genome-scale CRISPR gain-of-function screen in the same cell line showed sensitization upon CD48 overexpression.
A CRISPR screen in the AML cell line MOLM14 identified disruption of TNFRSF1B encoding TNFR2 as strongly conferring NK cell resistance. Interestingly, TNFRSF1B overexpression sensitized the MM cell line MM.1S to NK cell treatment in the gain-of-function screen. Integration with genomic data from patients with hematological malignancies revealed selective expression of TNFRSF1B in AML. Within AML, TNFRSF1B expression was enriched in myelomonocytic and monocytic subtypes as well as in MLL-rearranged cases represented by the MOLM14 cell line. Further dissection at the single-cell level revealed increased expression of TNFRSF1B with differentiation of AML cells along the monocytic lineage. Consistently, the less differentiated MOLM13 cell line established from the same patient as MOLM14 was resistant to NK cell killing, suggesting that a less differentiated phenotype of AML cells confers resistance to NK cell cytotoxicity through lack of TNFRSF1B expression.
Our data suggest that different lineages of hematological malignancies may be susceptible to NK cells through distinct mechanisms. In some cases, this heterogeneity is governed by lineage-specific expression of susceptibility genes, such as TNFRSF1B in monocytic AML. Particularly, our data identify a mechanism of NK cell evasion in less differentiated AML cells, suggesting potential for enhancing immune clearance of AML cells through differentiating therapies.
Lee:Kiadis Pharma: Consultancy, Equity Ownership, Membership on an entity's Board of Directors or advisory committees, Patents & Royalties, Research Funding. Mitsiades:Takeda: Other: employment of a relative ; Ionis Pharmaceuticals: Honoraria; Fate Therapeutics: Honoraria; Arch Oncology: Research Funding; Sanofi: Research Funding; Karyopharm: Research Funding; Abbvie: Research Funding; TEVA: Research Funding; EMD Serono: Research Funding; Janssen/Johnson & Johnson: Research Funding. Mustjoki:BMS: Honoraria, Research Funding; Novartis: Research Funding; Pfizer: Research Funding.

This feature is available to Subscribers Only
Sign In or Create an Account Close Modal