

Introduction: Pearson syndrome (PS) was originally reported as a sideroblastic anemia in infancy with vacuolization of marrow precursors and exocrine pancreas dysfunction. It is now recognized as a fatal multisystem mitochondrial disorder caused by single mitochondrial DNA deletions (SLSMDs) presenting with anemia. PS, Kearns-Sayre Syndrome (KSS) and progressive external ophthalmoplegia (PEO) form a continuous spectrum of disease associated with SLSMDs. There have been only a few systematic studies on PS.
Methods: We retrospectively reviewed hematological features and clinical course of 25 children with PS diagnosed between 1987 and 2019.
Results: Patients presented with normo/macrocytic transfusion-dependent anemia (n=25), failure to thrive (n=3), diarrhea (n=1), acidosis (n=1) and/or omphalocele/esophageal atresia (n=1) at a median age of 5 (0-31) months. A median hemoglobin, platelet count and neutrophil count were 6.5 (1.9-9.8) g/dl, 104 (31-300) G/L, and 0.9 (0.1-2.4) G/L, respectively. Bone marrow (n=24) was normo- (n=15) or hypocellular (n=9). Vacuoles in erythroid and myeloid precursors were observed in all patients, but ring sideroblasts were present in only 16 of 23 patients examined. Morphology can resemble Diamond-Blackfan anemia (DBA) because of erythroid hypoplasia (15/21). Dysplastic features are often observed including micromegakaryocytes. Lactic acid was elevated in most patients examined (14/18). Exocrine pancreas insufficiency at diagnosis was documented in 5 patients only. The detection of SLSMDs confirmed the diagnosis of PS in all patients.
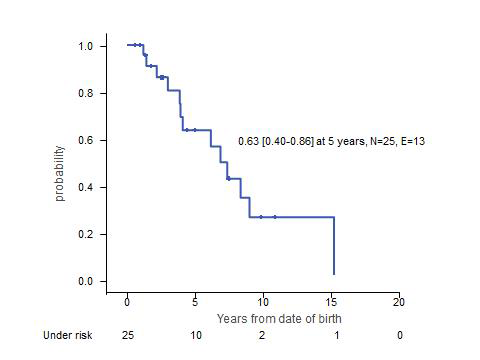
The median age at the time of the last follow-up was 47 (7 - 183) months. Among 11 patients with hematological follow-up for more than 3 years after diagnosis, 8 had spontaneous resolution of anemia at a median age of 28 (12-67) months, and 3 died at the age of 3, 6 or 8 years without hematological recovery. Clinical course was highly heterogeneous and various organ dysfunctions appeared. Renal tubulopathy/Fanconi syndrome (n=7) and cardiomyopathy/arrhythmia (n=5) were often fatal complications which developed at the median age of 32 and 45 months, respectively. Failure to thrive/short stature (n=13) and muscle hypotonia (n=9) were commonly observed. Other complications included pancreas insufficiency (n=7), liver dysfunction (n=4), endocrine dysfunctions (n=7), hearing loss (n=1), ophthalmoplegia (n=1), retinitis pigmentosa (n=1), cataract (n=1), ataxia (n=2) and encephalopathy (n=1). Thirteen patients died of acute metabolic acidosis with/without other complications (n=7), arrhythmia (n=2), respiratory failure (n=3) and liver/renal failure (n=1) at the median age of 50 (14-183) months. Two patients developed KSS and PEO-like phenotypes at the age 92 months and 19 months, respectively.
Summary: Anemia is generally the only presenting syndrome of PS. While the bone marrow morphology can resemble DBA or myelodysplastic syndrome, recognition of vacuolated myeloid/erythroid precursors lead to the correct diagnosis of PS in all cases. Other classical signs of PS, ring-sideroblasts and pancreas insufficiency, are often missing. Anemia spontaneously resolves in most patients surviving early childhood. However, PS is unexceptionally fatal (Figure), most patients succumb to metabolic acidosis and various forms of multi-organ failure. Since there is no effective therapy, the diagnosis of PS is one of the saddest news that pediatric hematologists have to break to parents of an anemic infant.
Niemeyer:Celgene: Consultancy.

This feature is available to Subscribers Only
Sign In or Create an Account Close Modal