

MDS and AML are separated according to the percentage of bone marrow blasts. Although this is an arbitrary threshold, it leads to different thinking and therapeutic strategies. Following the WHO classification for myeloid neoplasms an increasing number of specific genetic aberrations are incorporated in the classification, like t(15;17), t(8;21), inv(16) or NPM1 and CEBPA mutations for AML. Some of these are classified as AML even irrespective of blast counts.
Our aim was to stratify today´s AML and MDS patients by using genome sequencing data and a combination of machine learning techniques to identify the most prominent discriminative features, challenging the blast count as gold standard for discriminating AML from MDS.
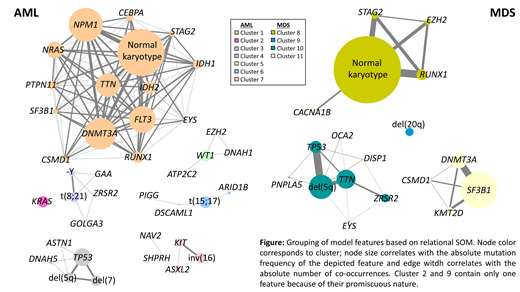
The analysis was based on a cohort of 1,292 patients (pts) morphologically diagnosed according to WHO classification: 591 AML and 701 MDS. Whole genome sequencing (WGS) was performed with 90x coverage for all samples to assess their mutational profiles. The Illumina tumor/unmatched normal workflow was used for variant calling. To remove the most frequent germline variants, each variant was queried against the gnomAD database, variants in non-coding regions and with global population frequencies >1% where excluded. The resulting dataset was filtered to exclude genes with a mutation frequency <1% resulting in a variant list assigned to 2,918 different genes. Additionally, the 10 most abundant cytogenetic aberrations in AML and MDS were used to train the model. The dataset was randomly divided into a training (90%) and validation (10%) set ensuring that all morphology based AML and MDS phenotypes according to WHO were present in both sets. Subsequently we applied LASSO regression to identify the features that optimize the classification accuracy of AML versus MDS. 500 models were built with 10-fold cross-validation to stratify the patients with an accuracy range from 66.7% - 95.2%. All the models differed slightly in their composition of selected features and, hence, the models with the highest accuracy (top 5%) were chosen, the selected features assessed and features that occurred in more than 50% of the models were kept to train a Naïve Bayes classifier. Using this final model to stratify patients of the validation cohort we achieved an accuracy of 83.7%. The model consisted of only 74 genetic markers, covering 8 cytogenetic aberrations and 66 affected genes. 26 of those genes belong to the COSMIC cancer gene list and of these 21 are well known markers included in myeloid screening panels. 40 genes and their variants (including rare polymorphisms, as well as variants or somatic mutations) have not been described in association with myeloid neoplasms so far. The model assigned 35/132 (27%) AML cases and 10/140 (7%) MDS cases to their respective counterparts. The median bone marrow blast count was 72% for concordant AML cases, 4% for concordant MDS cases, 44% for the falsely assigned AML cases and 8% for falsely assigned MDS patients, indicating that the blast count was lower in patients assigned to MDS instead of AML. However, the classification was based on the sample genetic background only and, hence, we grouped the model features based on relational self organizing maps (SOM) to identify entity-specific co-occurrence networks (7 AML and 4 MDS clusters, Figure 1). In AML the dominant cluster features were: normal karyotype with NPM1 mutation, t(8;21), t(15;17), inv(16), WT1 mutation, KRAS mutation, and TP53 mutation with co-occurring del(7) and del(5q). In MDS the 4 clusters were described by: del(5q), SF3B1 mutation, normal karyotype, and del(20q). All the misclassified cases showed a molecular profile that could be clearly associated with the ascertained entity-defining molecular features, explaining the divergent group assignment. Therefore, the molecular profiles indicate a considerable genetic similarity of the two diseases independent of the blast count. Considering the possible synergistic effects of co-occurring variants, the higher density of the interaction network in AML compared to MDS is noteworthy.
In conclusion, our study shows that a depth of information is hidden in the exome of myeloid neoplasms that can be uncovered by machine learning techniques and allows a genetic-based classification of AML and MDS. These techniques not only challenge the morphological classification, but also suggest that blast counts might not be the best parameter for treatment decision.
Meggendorfer:MLL Munich Leukemia Laboratory: Employment. Walter:MLL Munich Leukemia Laboratory: Employment. Haferlach:MLL Munich Leukemia Laboratory: Employment, Equity Ownership. Kern:MLL Munich Leukemia Laboratory: Employment, Equity Ownership. Haferlach:MLL Munich Leukemia Laboratory: Employment, Equity Ownership.

This feature is available to Subscribers Only
Sign In or Create an Account Close Modal