

Hematopoiesis, especially the early events of blood cell formation, has been mainly studied in bulk populations of cells and using progenitor colony formation assays; the familiar hierarchy of cell lineage differentiation and maturation, and associated regulatory factors have been inferred from these methods. However, these techniques often require extensive manipulation of cells, the exposure of cells to unphysiological conditions, aggregation of heterogeneous populations, and prior assumptions concerning cell function and gene expression. New single cell methodology avoids many of these potential experimental deficiencies.
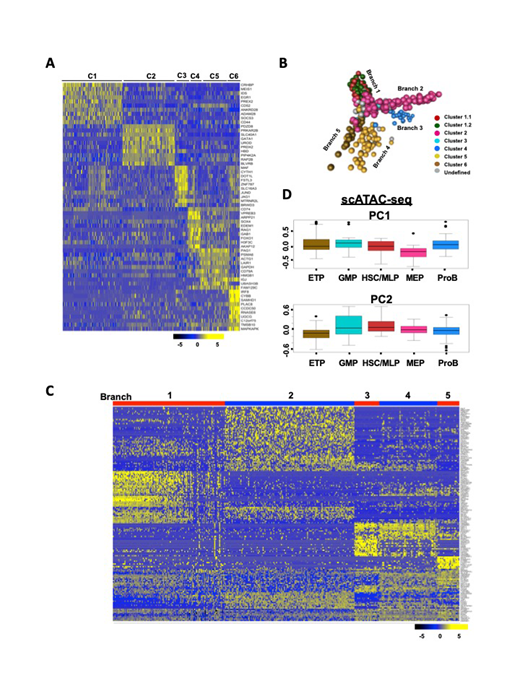
Here we have applied single-cell RNA-sequencing(scRNA-seq)to fresh human bone marrow CD34+cells: we profiled 391 single hematopoietic stem/progenitor cells (HSPCs) from four healthy donors by deep sequencing of individual cell transcriptomes. An average of 4560 protein-coding genes were detected per cell. Cells clustered into six distinct groups, which could be assigned to known HSPC subpopulations (Fig 1A), based on expression of lineage-specific genes. Lin-CD34+CD38+cells emerged as locally clustered cell populations (Clusters 2-6, including MEP, GMP, ETP and ProB), while Lin-CD34+CD38-cells formed a single cluster (HSC/MLP). Reconstruction of differentiation trajectories by transcription in single cells revealed four committed lineages derived from stem cell compartment. The earliest fate split separates MEPs from MLPs, which partition further into lymphoid, and granulocyte-monocyte progenitors (Fig 1B). The overall pattern differs from the classical hematopoietic model describing a single binary split between myeloid and lymphoid differentiation immediately downstream of multipotent cells. However, our data align well to recently published scRNA-seq data showing sequential commitment of stem cells to the lymphoid, erythroid/megakaryocytic, and finally myeloid lineages (Setty M, Nat Biotechnol2019; Pellin D, Nat Commun2019). We further examined trends in gene expression in each of the branches and found dynamic expression changes underlying cell fate during early lineage differentiation (Fig 1C). As confirmation, PCA plot of published single-cell assay for transposase-accessible chromatin (scATAC-seq) shows similar differentiation pattern. After projecting scATAC-seq data to our transcriptomic clusters' specific genes, MEP-dependent and myeloid/lymphoid-dependent genes were located on opposing sides of the PC1 with same direction (Fig 1D), indicating transcriptome and epigenome work on differentiation in concerted effort.
scRNA-seq provides opportunities for discovery and characterization at the molecular levels of early HSC differentiation and developmental intermediates, retrospectively, without the need to isolate purified populations. However, information inferred from scRNA-seq may be obscured due to missing reads and limited cell numbers. More cells would provide greater detail and higher resolution mapping.Given the low frequency of megakaryocyte progenitors within the CD34+cells as well as the neglected Lin-CD34-BM compartment, we could not fully resolve the separation and maturation of all lineages. Nonetheless, we found good coverage of cell types and a similar HSPC Atlas as other published studies (Velten L, Nat Cell Biol2017; Pellin D, Nat Commun2019)despite our limited numbers of starting cells. Our data accurately reflect the pattern of normal hematopoiesis, which may help to revise and refine characterization of hematopoiesis and provide a general reference framework to investigate the complexities of blood cell production at single-cell resolution - especially when cell numbers are limited, as from patient samples and in marrow failure syndromes.
Fig. 1scRNA-seq of human hematopoietic stem and progenitor cells. (A) Unsupervised hierarchical clustering of gene expression data for all cells. C1, HSC/MLP; C2, MEP; C3, GMP; C4, ProB; C5-C6, ETP. (B)Visualization of the HSPC continuum. Each ball represents one cell.(C) Large-scale shifts in gene expression during development of hematopoietic cells.Bars on top indicate locations of individual cells, colored by stages of development, along this developmental trajectory. (D) Projections of five transcriptomic gene modules onto PCA of scATAC-seq data (Buenrostro JD,Cell 2018). Each dot represents a transcriptional factor.
No relevant conflicts of interest to declare.

This feature is available to Subscribers Only
Sign In or Create an Account Close Modal