

Recent studies have examined the role of metabolism in pathogenic T cells in various diseases. The metabolic and bioenergetic characteristics of cells targeted by pathogenic T cells, such as gastrointestinal (GI) epithelial cells during immune-mediated disease processes are not known. Herein we explored the role of intestinal epithelial cell (IEC) metabolism on the severity of T cell mediated colitis including graft-versus-host disease (GVHD).
We first determined the bioenergetics of IECs following allogeneic hematopoietic cell transplantation (HCT). We utilized the well-characterized MHC disparate BALB/c→B6 GVHD model. Recipient B6 mice were lethally irradiated (10Gy), transplanted with 5×106 BM and 3x106 splenic T cells from either syngeneic B6 or allogeneic BALB/c donors. CD326+ IECs from recipient animals were harvested on day 7 and 21 post-HCT and assessed for their bioenegetics utilizing the Seahorse analyzer. The allogeneic IECs (allo-IECs) demonstrated dramatically reduced oxygen consumption rates (OCR) but similar extracellular acidification rates (ECAR), and increased OCR/ECAR ratios compared with syngeneic IECs (syn-IECs) (OCR; 44.70 vs 110 pmol/min, P<0.0001, ECAR; 34.43 vs 40.72 mpH/min, P=0.1832, OCR/ECAR; 1.12 vs 2.836, P<0.01). Because these data indicated profound defects in mitochondrial oxidative pathways, we next profiled mitochondrial tricarboxylic acid (TCA) cycle metabolite composition in IECs and kidney (as non-GVHD controls) harvested from naïve, syngeneic and allogeneic animals (day 7 and 21 post-HCT) in an unbiased and blinded manner with gas chromatography-mass spectrometry (GC-MS). Among the TCA cycle metabolites, only succinate significantly increased in IECs, but not kidneys, from allogeneic recipients (25.6 ± 5.65vs 0.00µM/µg protein, P<0.05).
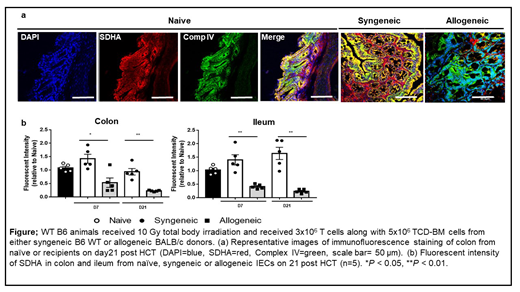
To determine the reasons for high levels of succinate, we next performed metabolic flux studies with 13C-glucose and 13C-glutamine, and found that the increased succinate levels were not secondary to anaplerosis. We therefore next hypothesized that the accumulated succinate in IECs was from reduced succinate dehydrogenase A (SDHA), a component of mitochondria complex II (MC II), that converts succinate to fumarate. Consistent with this hypothesis, SDHA was significantly decreased in allo-IECs compared to syn-IECs (P<0.05), which was additionally validated in a blinded manner by immunofluorescence (Figure), immunohistochemistry and immuno-gold staining of mitochondria by electron microscopy. Analysis of IECs harvested from recipients following non-irradiated B6→B6D2F1, chemotherapy conditioned BALB/c→B6, and irradiated MHC matched mHA mismatched C3H.SW→B6 GVHD models, as well as in a CD4+CD45RBhigh T-cell→RAG1−/− model of inflammatory bowel disease (IBD), and an anti-CTLA-4 antibody augmented DSS colitis model (checkpoint-inhibitor colitis), all demonstrated reduction of MC II, SDHA, and an increase in the level of succinate.
We next explored the in vivo functional relevance of SDHA in GI GVHD utilizing multiple approaches. Specifically, when Sdhaf1−/− B6 (30% of SDHA activity, P<0.01), bone marrow chimera (WT B6→Sdhaf1−/−, P<0.05) or WT animals treated with multiple chemical SDH inhibitors from day 0 to 21 post allo-HCT (itaconate; 2.5g/kg, P<0.0001, malonate; 5g/kg, P<0.001 or Atpenin A5; 9µg/kg, P<0.05) were used as recipients, all demonstrated significantly greater GI GVHD and mortality compared with allo-control animals. We furthermore generated IEC specific SDHA KO mice (villin-Cre+SDHAfl/fl), which when utilized as allogeneic recipients, they demonstrated significantly greater mortality and GI GVHD (P<0.0001).
Cellular and biochemical mechanistic studies demonstrated the requirement of T cell contact with IECs, and a critical role for perforin (Prf)/ granzyme B (GB) in the breakdown and reduction of SDHA. The mechanisms for SDHA reduction were validated in vivo by transferring Prf−/− T cells into allogeneic BALB/c (GVHD) or RAG1−/−B6 (IBD) mice.
Our data demonstrate that MC II component SDHA in IECs is a critical metabolic checkpoint that regulates severity of intestinal colitis caused by pathogenic allo- or auto-reactive T cells and thus provide seminal insights into GI GVHD, IBD and checkpoint-inhibitor colitis.
No relevant conflicts of interest to declare.

This feature is available to Subscribers Only
Sign In or Create an Account Close Modal