

In previous studies of recurrently amplified 1q21 genes in multiple myeloma (MM), we identified ILF2 (Interleukin Enhancer Binding Factor 2) as a key modulator of the DNA repair pathway, which promotes adaptive responses to genotoxic stress in a dose-dependent manner, explaining why 1q21 patients benefit less from high-dose chemotherapy than non-1q21 patients do (Marchesini, Cancer Cell 2017). These findings prompted us to develop strategies for blocking ILF2 signaling to enhance the effectiveness of available DNA-damaging agent-based treatments. Given that ILF2 is selectively overexpressed in 1q21 MM cells and is not easily amenable to small-molecule or antibody depletion, we collaborated with IONIS Pharmaceuticals to develop antisense nucleotides targeting ILF2 (ILF2 ASOs). To exclude on-target toxicities that could arise from ILF2 inhibition, we injected 14 different ASOs targeting mouse ILF2 into male Balb/c mice. Of the 14 ILF2 ASOs we tested, 6 did not induce either notable histopathological findings or hematological and biochemical alterations, which suggests that ILF2 inhibition is well-tolerated in normal tissues. Thus, ILF2 ASOs were used for functional validation studies in myeloma.
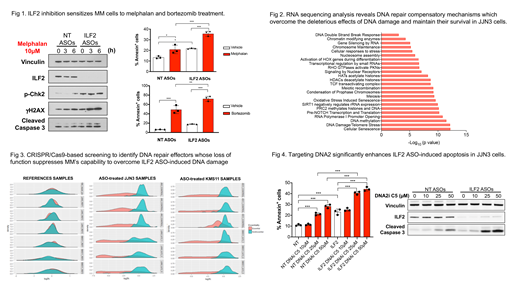
Consistent with our previous work using ILF2-targeting shRNAs, we observed that ILF2 ASO-induced ILF2 depletion resulted in significantly inhibited cell proliferation, increased ATM/Chk2 pathway activation, γH2AX accumulation, and caspase 3-mediated apoptosis in KMS11 and JJN3 cells and sensitized these cells to melphalan, bortezomib and olaparib treatment (Fig 1). However, whereas KMS11 cells had a high level of DNA damage activation and a significantly higher apoptosis rate after more than 2 weeks of ILF2 ASO treatment, JJN3 cells overcame ILF2 ASO-induced DNA damage activation and apoptosis and became resistant to ILF2 ASO treatment. To gain insights into the molecular mechanisms by which MM cells can overcome ILF2 ASO-induced DNA damage activation, we subjected ILF2 ASO-treated KMS11 and JJN3 cells to RNA sequencing analysis at early and late treatment times. We found that the genes that were significantly downregulated in JJN3 but not KMS11 cells treated with ILF2 ASOs for more than 2 weeks as compared with those treated for 1 week were mostly involved in the regulation of the DNA damage response (Fig 2). These findings suggest that MM cells can activate compensatory mechanisms to overcome the deleterious effects of DNA damage and survive.
To identify DNA repair effectors whose loss of function suppresses 1q21 MM cells' capability to overcome ILF2 ASO-induced DNA damage, we performed a CRISPR/Cas9 screening using a pool of single-guide RNAs (sgRNAs) targeting 196 genes involved in the DNA damage response. Using the drugZ algorithm to assess differences in the representation of all sgRNAs between cells treated with NT or ILF2 ASOs for 3 weeks (Fig 3), we found that sgRNAs targeting the DNA replication helicase/nuclease 2 (DNA2) were among the most significantly depleted sgRNAs in ILF2 ASO-treated JJN3 cells, whereas sgRNAs targeting the Fanconi anemia core complex-associated protein 24 (FAAP24) were significantly depleted in ILF2 ASO-treated KSM11 cells. Using the DNA2 inhibitor C5, we further validated that targeting DNA2 significantly enhances ILF2 ASO-induced apoptosis in JJN3 cells (Fig 4). Functional validation experiments using inducible sgRNAs are ongoing to evaluate whether the inhibition of DNA2 or FAAP24 is a synthetic lethal approach to targeting 1q21 MM cells in the setting of therapies with DNA-damaging agents.
Collectively, our study demonstrates that ILF2 ASO therapy may be exploited to optimize the use of DNA-damaging agents in patients with 1q21 MM.
Garcia-Manero:Amphivena: Consultancy, Research Funding; Helsinn: Research Funding; Novartis: Research Funding; AbbVie: Research Funding; Celgene: Consultancy, Research Funding; Astex: Consultancy, Research Funding; Onconova: Research Funding; H3 Biomedicine: Research Funding; Merck: Research Funding. Colla:Abbvie: Research Funding; Amgen: Research Funding; IONIS: Other: Intellectual property and research material IONIS).

This feature is available to Subscribers Only
Sign In or Create an Account Close Modal