

In this issue of Blood, provide new insights into potential mechanisms for thromboinflammatory complications associated with red blood cell (RBC) transfusions. By using leukoreduced RBC units to isolate RBC microvesicles (RBC-MVs), they document that RBC-MVs activate factor IX (FIX) via 2 distinct pathways: (1) the canonical intrinsic pathway in which activated FXII (FXIIa) activates FIX in an FXI-dependent manner and (2) a noncanonical pathway in which plasma kallikrein directly activates FIX, which ultimately results in thrombin generation.1
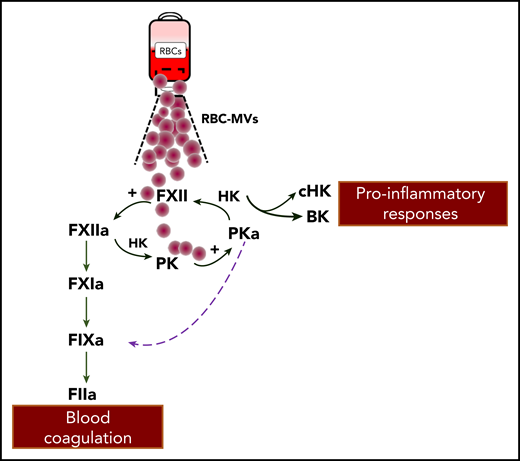
RBC-MVs interact with the contact system to activate coagulation FIX. RBC-MVs that accumulate during storage of RBC units can directly activate FXII and prekallikrein (PK). FXII activation of PK forms plasma kallikrein (PKa) that reciprocally activates FXII and liberates bradykinin (BK) from high molecular weight kininogen (HK). In the canonical pathway proposed by Noubouossie et al, FXIIa activation of FXI leads to activated FIX. In an alternative pathway, PKa directly activates FIX. The sum of these activities leads to a series of proteolytic reactions and ultimately, to the generation of thrombin (FIIa). Generated BK can influence vascular smooth muscle tone, vascular permeability, and leukocyte functions.
RBC-MVs interact with the contact system to activate coagulation FIX. RBC-MVs that accumulate during storage of RBC units can directly activate FXII and prekallikrein (PK). FXII activation of PK forms plasma kallikrein (PKa) that reciprocally activates FXII and liberates bradykinin (BK) from high molecular weight kininogen (HK). In the canonical pathway proposed by Noubouossie et al, FXIIa activation of FXI leads to activated FIX. In an alternative pathway, PKa directly activates FIX. The sum of these activities leads to a series of proteolytic reactions and ultimately, to the generation of thrombin (FIIa). Generated BK can influence vascular smooth muscle tone, vascular permeability, and leukocyte functions.
Approximately 12 million units of stored RBCs are transfused annually in the United States alone.2 In vivo, RBCs have evolved to transport oxygen by redox enzymatic reactions until they become damaged and are rapidly removed from the circulation. However, these mechanisms are no longer operative when RBCs are stored in a blood bank. The resultant loss or degradation of RBC components is collectively referred to as “storage lesion” and accounts for the limited shelf life of RBC units (up to ∼7 weeks). Elements of the RBC storage lesion include oxidative damage, enzymatic malfunction, and structural abnormalities, including the formation of RBC-MVs. These MVs are derived from parent RBCs, are submicron in size, and have been described to have thrombin-generating potential on the basis of clinical observations, in vitro experiments, and animal model studies.
The mechanisms by which RBC-MVs exert procoagulant activity have been the focus of previous research. Fisher et al3 reported that RBC-MVs engage in heterotypic interactions with monocytes and found that tissue factor expression in monocytes (and plasma) increased after exposure to RBC-MVs. RBC-MVs were also shown to promote phenotypic changes in platelets, wherein enhanced surface expression of P-selectin and activated GPIIb/IIIa was noted when platelets were exposed to RBC-MVs.3,4 In contrast to their parental cells, the majority of RBC-MVs expose negatively charged phospholipids (mainly phosphatidylserine), which are essential for the assembly of the tenase and prothrombinase complexes.5 One question that arises is which components of the coagulation system initiate and propagate RBC-MV–mediated effects? Using calibrated automated thrombography, Rubin et al6 showed that thrombin generation in response to RBC-MVs was not influenced by the absence of FVII, was mildly decreased by FXII deficiency, and was severely impaired by the absence of FVIII or FIX. Notably, there was no thrombin generation in FXI-deficient plasma or after treatment with an FXIa-blocking antibody, suggesting that RBC-MVs have FXI-dependent procoagulant properties.6 These findings set the stage for the Noubouossie study to critically assess how RBC-MVs initiate coagulation.
Using purified systems to assess coagulation factor activities and thrombin generation, the investigators provide convincing evidence that compared with healthy plasma, thrombin generation induced by RBC-MVs in FXI- or FXII-deficient plasma was reduced but not abolished, as previously proposed in the Rubin et al6 study. In contrast, RBC-MV–mediated thrombin generation was abrogated in FIX-deficient plasma. This supports the notion that although FXI and FXII contribute to thrombin generation induced by RBC-MVs, FXI- and FXII-independent pathways seem to exist, whereas FIX is indispensable to the process. To determine which zymogens of the intrinsic pathway are directly activated by RBC-MVs, the investigators measured coagulant activity for each of these factors. They observed that RBC-MVs generated significant levels of FXIIa but not FIXa or FXIa, and they concluded that (1) initiation of thrombin generation occurs via the intrinsic pathway upstream of FIX and (2) given that residual thrombin generation was seen in FXI- and FXII-deficient plasmas, there is an alternate pathway for FIX activation by enzymes of the contact system.
In search of candidate FIX activators, investigators made the important observation that plasma kallikrein itself promotes FIXa formation. FXII and plasma prekallikrein reciprocally activate each other and result in liberation of bradykinin.7 Therefore, FXIIa generated by RBC-MVs can partly divert toward kallikrein formation, which amplifies FIX activation. The authors pursue this scenario by showing that RBC-MV–induced thrombin generation is abolished only in the presence of both corn trypsin inhibitor and soybean tryspin inhibitor. Collectively, these findings show that after contact activation by RBC-MVs, 2 distinct pathways proceeding through FXIIa- and kallikrein-mediated activation of FIX support their procoagulant effects (see figure). Moreover, the authors report that heating RBC-MVs (albeit at nonphysiologic temperatures) inhibited thrombin generation but not prothrombinase activity, which suggests that a heat-sensitive factor is the rate-limiting step in the initiation of thrombin generation by RBC-MVs.
These findings add to our understanding of the complex interactions between cell-derived MVs and components of the coagulation system. However, several questions remain unanswered. First, it is not clear how RBC-MVs activate FXII. Is RBC-MV–induced FXII activation dependent on membrane properties of MVs or on the cargo contained in the structures? Second, are the 2 pathways leading to FIX activation uncoupled? Or do they proceed simultaneously, and if so, what is their selective contribution to thrombin generation in the presence of RBC-MVs? With regard to clinical translation of these findings, the in vivo relevance of RBC-MV–mediated procoagulant responses has yet to be established, especially in light of recent clinical studies that report no association between blood transfusions and thrombosis outcomes.8 Perhaps certain high-risk patient populations such as polytrauma cases (in whom massive transfusion protocols are commonly implemented and the burden of RBC-MVs may rise) can be studied for potential thromboinflammatory sequelae. These key questions will likely be investigated in future studies.
In spite of these remaining questions, the study by Noubouossie et al provides significant insight about the potential of cell-derived MVs to elicit thrombotic and immunomodulatory responses. In the past 15 years, there has been a revival of interest in the contact system as more biologic substances have been recognized to support FXII activation in vivo, as novel FXII functions have emerged, and as animal and human studies have shown that the contact system is dispensable for hemostasis but contributes to thrombosis.9,10 The study by Noubouossie et al adds to the growing list of potentially relevant FXII activators and sets the stage for future experimental studies to address the therapeutic potential of inhibiting the contact system as a strategy for preventing and managing complications for recipients of RBC transfusions.
Conflict-of-interest disclosure: The author declares no competing financial interests.
