

Key Points
Microvesicles in stored RBC units activate the contact pathway, resulting in both FXIIa- and kallikrein-mediated activation of FIX.
These pathways are potential targets to prevent thrombotic or inflammatory complications of red cell transfusion.

Abstract
Storage lesion–induced, red cell–derived microvesicles (RBC-MVs) propagate coagulation by supporting the assembly of the prothrombinase complex. It has also been reported that RBC-MVs initiate coagulation via the intrinsic pathway. To elucidate the mechanism(s) of RBC-MV–induced coagulation activation, the ability of storage lesion–induced RBC-MVs to activate each zymogen of the intrinsic pathway was assessed in a buffer system. Simultaneously, the thrombin generation (TG) assay was used to assess their ability to initiate coagulation in plasma. RBC-MVs directly activated factor XII (FXII) or prekallikrein, but not FXI or FIX. RBC-MVs initiated TG in normal pooled plasma and in FXII- or FXI-deficient plasma, but not in FIX-deficient plasma, suggesting an alternate pathway that bypasses both FXII and FXI. Interestingly, RBC-MVs generated FIXa in a prekallikrein-dependent manner. Similarly, purified kallikrein activated FIX in buffer and initiated TG in normal pooled plasma, as well as FXII- or FXI-deficient plasma, but not FIX-deficient plasma. Dual inhibition of FXIIa by corn trypsin inhibitor and kallikrein by soybean trypsin inhibitor was necessary for abolishing RBC-MV–induced TG in normal pooled plasma, whereas kallikrein inhibition alone was sufficient to abolish TG in FXII- or FXI-deficient plasma. Heating RBC-MVs at 60°C for 15 minutes or pretreatment with trypsin abolished TG, suggesting the presence of MV-associated proteins that are essential for contact activation. In summary, RBC-MVs activate both FXII and prekallikrein, leading to FIX activation by 2 independent pathways: the classic FXIIa-FXI-FIX pathway and direct kallikrein activation of FIX. These data suggest novel mechanisms by which RBC transfusion mediates inflammatory and/or thrombotic outcomes.
Introduction
Microvesicles (MVs) are submicron membrane-derived vesicles released by many cells upon stimulation or during apoptosis.1 MVs express membrane proteins derived from their cell of origin.2 Some MVs are considered to be procoagulant because they express tissue factor and/or a phosphatidylserine (PS) surface that supports the assembly of enzymatic coagulation complexes.3
Red blood cells (RBCs) stored in the blood bank undergo structural and metabolic changes that are collectively known as the red cell storage lesion.4 These changes include the release of RBC-MVs.5,6 Artificially generated RBC-MVs have been shown to activate coagulation independent of tissue factor.7,8 Storage lesion–induced MVs are of interest because they are infused into patients along with blood components. Furthermore, multiple retrospective studies have reported an association between RBC transfusion and thrombosis,9-14 although this has been disputed.15 Previous studies have demonstrated that storage lesion–induced RBC-MVs amplify tissue factor–initiated thrombin generation (TG) in plasma,16 an effect that is likely explained by PS exposure.17,18 Although coagulation activation by storage lesion–induced RBC-MVs is independent of the extrinsic pathway,16,19 these MVs also initiate TG in normal plasma containing corn trypsin inhibitor (CTI), a potent inhibitor of FXIIa, as well as in FXII-deficient plasma, supporting the notion that FXII(a) may not be necessary.16 However, TG is attenuated in the presence of an inhibitory anti-FXI antibody, indicating dependence on FXI.16,19
To further explore the mechanism by which RBC-MVs initiate coagulation, we used purified systems to examine their interactions with coagulation factors of the intrinsic pathway. We demonstrate that storage lesion–induced RBC-MVs activate FXII and plasma prekallikrein (PK), leading to FIX activation via 2 distinct pathways: (1) the canonical intrinsic pathway in which FXIIa indirectly activates (FIX) via FXI activation; and (2) a noncanonical pathway in which kallikrein (PKa) directly activates FIX leading to TG.
Methods
Materials
Unless otherwise stated, the buffer used for functional assays in this study contained 20 mM N-2-hydroxyethylpiperazine-N′-2-ethanesulfonic acid (HEPES), 150 mM NaCl, 0.1% polyethylene glycol 8000, 2 mM CaCl2, 1.2 mM MgCl2, and 20 µM ZnCl2 at pH 7.4. Unilamellar synthetic lipid vesicles (15% PS, 41% phosphatidylethanolamine [PE], and 44% phosphatidylcholine [PC]) were synthesized as previously described.20 CTI was prepared as previously described.21 The 5G9 antibody (which inhibits the tissue factor-FVIIa complex) was a gift from Wolfram Ruf. Full-length exopolyphosphatase was expressed in Escherichia coli and purified as previously described.22 Heparinase-1, -2, and -3 and chondroitinase ABC were from Jian Liu (UNC Eshelman School of Pharmacy). All other materials were purchased from commercial suppliers, as described in supplemental Methods, available on the Blood Web site.
Isolation and characterization of RBC-MVs
The American Red Cross (Carolinas Region) prepared leukoreduced RBC concentrate units. The units were stored and handled until expiration (42 days) in the blood bank of the VA Medical Center (Durham, NC), in accordance with American Association of Blood Banks and US Food and Drug administration standards. RBC-MV isolation was performed within 2 days after expiration. In addition, a few in-date “younger” RBC units that were released by the blood bank and not ultimately transfused were available for study. These units were then maintained in blood bank storage conditions and subsampled at different time points for MV isolation. An aliquot of RBC concentrates was transferred into a 50-mL plastic conical tube and centrifuged twice at 2500 g for 15 minutes each, as recommended by the International Society on Thrombosis and Haemostasis.23 Detailed methods used to characterize isolated RBC-MVs are provided in supplemental Methods. In brief, isolated RBC-MVs were visualized after negative staining using transmission electron microscopy. Dynamic light scattering was used to confirm that isolated RBC-MVs contained only submicron MVs before nanoparticle tracking analysis (NTA) was used for precise sizing and enumeration. PS expression on RBC-MVs was tested by using a commercially available prothrombinase activity method (Zymuphen MP-Activity). RBC-MVs were pelleted by centrifugation at 20 000 g for 30 minutes, washed, and resuspended in phosphate-buffered saline for subsequent functional assays.
TG assay
MV-free normal pooled, or factor-deficient plasmas were prepared as indicated in supplemental Methods. TG assays were performed as previously described,24 with some modifications. In brief, 20 µL buffer, kaolin, human PKa, or washed RBC-MVs were added to 80 µL of MV-free plasma in a black 96-well microplate. Then, 20 µL of a solution containing 90 mM CaCl2, 2.496 mM Z-Gly-Gly-Arg-AMC fluorogenic thrombin substrate. and 2.4 μM synthetic lipid vesicles dissolved in 20 mM N-2-hydroxyethylpiperazine-N′-2-ethanesulfonic acid–buffered saline was added. Substrate hydrolysis was monitored over time in a Biotek Synergy H1M fluorometer. TG curves were obtained by plotting the first derivative of fluorescence read as a function of time.
Factor XII activation assay
To assess FXII activation, 20 µL of buffer, kaolin, synthetic lipid vesicles, or washed RBC-MVs was added to 80 µL of 375 nM human FXII dissolved in buffer. Twenty microliters of fluorogenic substrate (Boc-Gln-Gly-Arg-AMC; 100 µM final concentration) was added, and FXIIa generation was monitored for 150 minutes by measuring substrate cleavage–induced fluorescence at 360/460 nm excitation/emission wavelengths. Results are presented as relative fluorescence units (RFUs) over time, or as the difference in RFUs at 2 hours of incubation minus the initial reading.
PK activation assay
To assess PK activation, 20 µL of buffer, kaolin, synthetic lipid vesicles or washed RBC-MVs was added to 80 µL of 580 nM human PK dissolved in buffer in the presence of 670 nM human high molecular weight kininogen (HK). Twenty microliters of fluorogenic substrate (Boc-Gln-Gly-Arg-AMC; 100 µM final concentration) was added, and PKa generation was monitored for 150 minutes by measuring substrate cleavage–induced fluorescence at 360/460 nm excitation/emission wavelengths. Results are presented as RFUs over time or as the difference in RFUs at 2 hours of incubation minus the initial reading.
Factor XI activation assay
To assess FXI activation, 20 µL of buffer, FXIIa, synthetic lipid vesicles, or washed RBC-MVs was added to 80 µL of 30 nM human FXI dissolved in buffer in the presence of 150 nM human HK. Twenty microliters of fluorogenic substrate (Boc-Gln-Gly-Arg-AMC; 100 µM final concentration) was added, and FXIa generation was monitored for 150 minutes at 37°C by measuring substrate cleavage–induced fluorescence at 360/460 nm excitation/emission wavelengths. Results are presented as RFUs over time or as the difference in RFUs at 2 hours of incubation minus the initial reading.
Factor IX activation assay
For FIX activation, 20 µL of the activator (kaolin, human PKa, FXIIa, plasmin, FXIa, and synthetic sPLs, washed RBC-MVs or buffer control) was incubated for 1 hour at 37°C with 80 µL of 100 nM human FIX dissolved in buffer. In some experiments, 570 nM human PK and 680 nM human HK were added to FIX in the buffer before addition of the activator. After incubation, human antithrombin (AT; 2.5 µM) and unfractionated heparin (1 U/mL) were added to the reaction and incubated for 30 additional minutes at 37°C. Then, the reaction was quenched by adding 20 µM of a cocktail of serine protease inhibitors (tosyl-lysyl-chloromethyl ketone, tosyl-phenyl-chloromethyl ketone, dansyl-Glu-Gly-Arg chloromethyl ketone, Phe-Pro-Arg chloromethyl ketone, and phenylmethylsulfonyl fluoride). FIXa generation was assessed by measuring the concentration of FIXa-AT, by using an in-house sandwich enzyme-linked immunosorbent assay (ELISA) that captures FIXa and detects AT (details in supplemental Methods). For the ELISA standard, 100 nM human FIXa was incubated for 1 hour at 37°C with 2.5 µM human AT in the presence of 1 U/mL unfractionated heparin. Results are expressed as concentrations of FIXa-AT complexes.
Results
Characterization of storage lesion–induced RBC-MVs
Isolated RBC-MVs were imaged by electron microscopy as individual particles or aggregates of spherical vesicles of various sizes (Figure 1A). Dynamic light-scattering analysis showed that isolated RBC-MVs were <1 µm in diameter (supplemental Figure 1A). NTA revealed that, although the size of RBC-MVs varied from 10 to 700 nm, the majority (97%–99%) had a diameter ≤200 nm (supplemental Figure 1B). RBC-MVs were strongly positive for surface expression of PS, comparable to that of 4 µM synthetic lipid vesicles containing 15% PS (Figure 1B). The concentration of stored RBC-MVs increased gradually with duration of storage from an average of 1.15 × 1012 MVs/mL at days 15 to 17 to 4.54 × 1012 MVs/mL at day 42 and 5.68 × 1012 MVs/mL at day 45 (Figure 1C). However, the size distribution of RBC-MVs did not vary significantly during the storage time (Figure 1D).
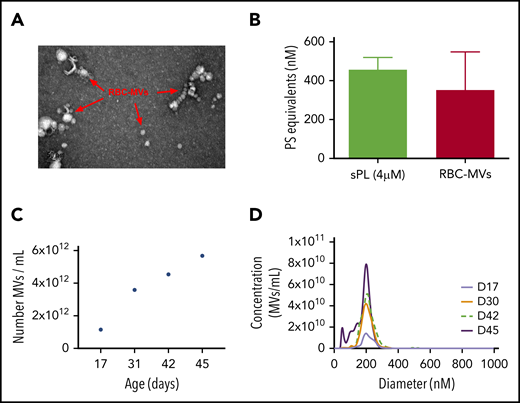
Structural characteristics of RBC-MVs. (A) RBC-MVs visualized by electron microscopy after negative staining. (B) PS expression on RBC-MVs or sPL vesicles (15% PS, 41% PE, 44% PC) measured using a prothrombinase activity assay. (C) Representative donor sample of the number of MVs per milliliter (of liquid fraction of transfusion unit) over storage time in days. (D) Size distribution and concentration of MVs per milliliter at the day of storage noted, from a representative donor sample.
Structural characteristics of RBC-MVs. (A) RBC-MVs visualized by electron microscopy after negative staining. (B) PS expression on RBC-MVs or sPL vesicles (15% PS, 41% PE, 44% PC) measured using a prothrombinase activity assay. (C) Representative donor sample of the number of MVs per milliliter (of liquid fraction of transfusion unit) over storage time in days. (D) Size distribution and concentration of MVs per milliliter at the day of storage noted, from a representative donor sample.
TG initiated by RBC-MVs in plasma is independent of the extrinsic pathway
Addition of RBC-MVs to recalcified normal pooled plasma (NPP) triggered TG in a dose-dependent manner (supplemental Figure 2A). In contrast, no TG was observed in recalcified NPP supplemented with up to 400 µM synthetic lipid vesicles (supplemental Figure 2B). Stored RBC-MV–induced TG gradually increased with duration of storage (Figure 2A). When the RBC-MV concentration was plotted against peak TG, we observed a positive linear correlation that reflected the duration of storage (supplemental Figure 2C). These data indicate that TG increases with the duration of RBC storage, primarily as a result of an increase in RBC-MVs. Robust TG induced by RBC-MVs was still observed in the presence of inhibitory antibodies to tissue factor (clone HTF1) or tissue factor-FVIIa complex (clone 5G9) or in FVII-deficient plasma (Figure 2B). Furthermore, tissue factor activity was undetectable on RBC-MVs (Figure 2C). Collectively, these results indicate that RBC-MVs initiate TG in plasma, independent of the extrinsic coagulation pathway, consistent with previous reports.16,19
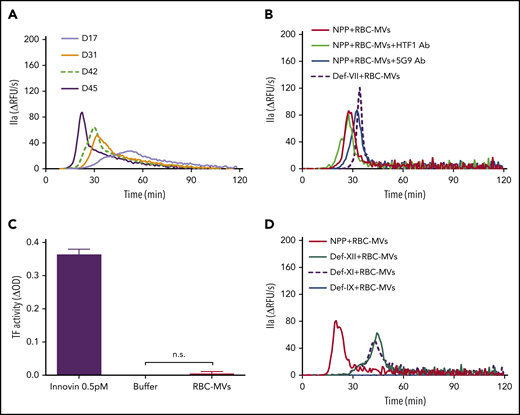
Procoagulant activity of RBC-MVs assessed using TG or an MV tissue factor activity assay. (A) TG in recalcified MV-free NPP in MVs from the indicated day (D) of storage, from a representative donor sample. (B) TG was performed in NPP in the absence (red) or presence (green) of anti-human tissue factor (HTF1 Ab), anti-human tissue factor-FVIIa complex (5G9 Ab; blue), or MV-free plasma deficient in FVII (def-VII; dashed dark purple). (C) Measurement of tissue factor activity on RBC-MVs. Innovin (0.5 pM) was used as the positive control and buffer as the negative control. Data are presented as the mean ± standard deviation of 3 independent experiments. (D) TG performed in MV-free plasma deficient in FXII (def-XII; green), deficient in FXI (def-XI; dashed dark purple), or deficient in FIX (def-IX; blue), compared with NPP (red). Data are representative of 3 independent experiments.
Procoagulant activity of RBC-MVs assessed using TG or an MV tissue factor activity assay. (A) TG in recalcified MV-free NPP in MVs from the indicated day (D) of storage, from a representative donor sample. (B) TG was performed in NPP in the absence (red) or presence (green) of anti-human tissue factor (HTF1 Ab), anti-human tissue factor-FVIIa complex (5G9 Ab; blue), or MV-free plasma deficient in FVII (def-VII; dashed dark purple). (C) Measurement of tissue factor activity on RBC-MVs. Innovin (0.5 pM) was used as the positive control and buffer as the negative control. Data are presented as the mean ± standard deviation of 3 independent experiments. (D) TG performed in MV-free plasma deficient in FXII (def-XII; green), deficient in FXI (def-XI; dashed dark purple), or deficient in FIX (def-IX; blue), compared with NPP (red). Data are representative of 3 independent experiments.
TG initiated by RBC-MVs in plasma is dependent on FIX
To determine whether TG induced by RBC-MVs was dependent on activation of the intrinsic pathway, RBC-MVs were resuspended in recalcified MV-free plasma deficient in FXII, FXI, or FIX. In the absence of RBC-MVs, no TG was detected in any of these factor-deficient plasmas after recalcification and supplementation with 4 µM synthetic lipid vesicles. When compared with NPP, TG induced by RBC-MVs in FXII- or FXI-deficient plasma was delayed and reduced, but not abolished (Figure 2D), even when CTI, a FXIIa inhibitor, was added to FXII-deficient plasma (supplemental Figure 3A). Likewise, a delayed and markedly attenuated TG was observed in NPP in the presence of an inhibitory anti-human FXI antibody (300 nM final concentration; supplemental Figure 3B). In contrast, RBC-MV–induced TG was abolished in FIX-deficient plasma (Figure 2D). These data demonstrate that TG induced by RBC-MVs is driven by the intrinsic coagulation pathway. Furthermore, they suggest that although FXII and FXI contribute to TG induced by RBC-MVs, FXII/FXI-independent pathways also appear to exist, and FIX is absolutely essential for TG.
RBC-MVs directly activate FXII and plasma PK but not FXI or FIX
Next, we sought to determine which zymogens of the intrinsic pathway are directly activated by RBC-MVs. Physiological concentrations of FXII, PK, FXI, or FIX were incubated in buffer, with or without RBC-MVs. HK was added to the reaction for PK or FXI activation, because these 2 zymogens circulate in plasma bound to HK, a cofactor that may affect their activation and/or enzymatic activity.25,26 RBC-MVs generated FXIIa from added FXII (Figure 3A). RBC-MVs also promoted the activation of PK (Figure 3B). In contrast, RBC-MVs did not directly promote activation of FXI (Figure 3C) or FIX (Figure 3D). Incubation of PK, FXI, or FIX with 10 µM synthetic phospholipid vesicles did not activate any of these coagulation factors (Figure 3). Overall, these results indicate that RBC-MVs initiate TG in plasma by promoting the activation of both FXII and PK.
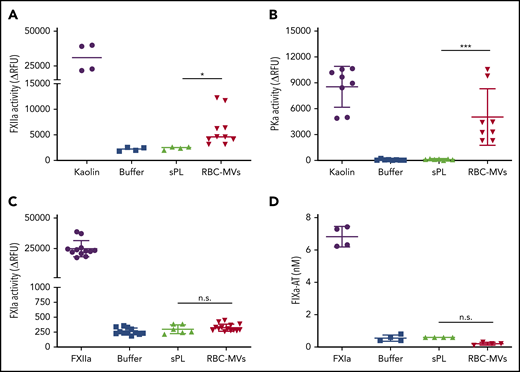
Assessment of direct activation of enzymes of the intrinsic pathway of coagulation by RBC-MVs or sPL vesicles in purified systems. (A) FXIIa activity generated after incubation of MVs (red) with 375 nM purified human FXII for 2 hours at 37°C. (B) Kallikrein activity generated after incubation of MVs with 580 nM purified human PK and 670 nM HK for 2 hours at 37°C. (C) FXIa activity generated after incubation of MVs with 30 nM purified human FXI and 150 nM HK for 2 hours at 37°C. (D) FIXa-AT complexes formed after incubation of MVs with 100 nM purified human FIX for 1 hour at 37°C, then addition of human AT (2.5 µM) and 1 U/mL unfractionated heparin. Enzyme activity was measured using fluorogenic substrate: 10 µg/mL kaolin (A-B), 10 nM FXIIa (C), or 0.5 nM FXIa as the positive control (purple) or buffer (blue) as the negative control (D). Lipid sPL vesicles (green) contained 15% PS, 41% PE, and 44% PC. FIXa-AT was measured with an in-house ELISA, capturing FIX and detecting AT. Data are expressed as the mean ± standard deviation of at least 4 independent experiments run as duplicates. Each experimental condition was compared with buffer using Dunnett’s multiple-comparisons test. *P < .05; ***P < .001.
Assessment of direct activation of enzymes of the intrinsic pathway of coagulation by RBC-MVs or sPL vesicles in purified systems. (A) FXIIa activity generated after incubation of MVs (red) with 375 nM purified human FXII for 2 hours at 37°C. (B) Kallikrein activity generated after incubation of MVs with 580 nM purified human PK and 670 nM HK for 2 hours at 37°C. (C) FXIa activity generated after incubation of MVs with 30 nM purified human FXI and 150 nM HK for 2 hours at 37°C. (D) FIXa-AT complexes formed after incubation of MVs with 100 nM purified human FIX for 1 hour at 37°C, then addition of human AT (2.5 µM) and 1 U/mL unfractionated heparin. Enzyme activity was measured using fluorogenic substrate: 10 µg/mL kaolin (A-B), 10 nM FXIIa (C), or 0.5 nM FXIa as the positive control (purple) or buffer (blue) as the negative control (D). Lipid sPL vesicles (green) contained 15% PS, 41% PE, and 44% PC. FIXa-AT was measured with an in-house ELISA, capturing FIX and detecting AT. Data are expressed as the mean ± standard deviation of at least 4 independent experiments run as duplicates. Each experimental condition was compared with buffer using Dunnett’s multiple-comparisons test. *P < .05; ***P < .001.
Purified kallikrein directly activates FIX in buffer and initiates TG in plasma in a FIX-dependent manner
RBC-MVs did not trigger TG in FIX-deficient plasma and did not directly activate FIX in buffer, indicating that initiation of TG must occur via the intrinsic pathway upstream of FIX. However, residual TG was observed when RBC-MVs were resuspended in recalcified FXII- or FXI-deficient plasma, suggesting the possibility of an alternate pathway for FIX activation by activated enzymes of the contact system. We hypothesized that FXIIa or PKa could activate FIX independently of FXI. Furthermore, since FXIIa can activate plasminogen, we evaluated whether plasmin might explain this observation. Therefore, the ability of purified human FXIIa, PKa, or plasmin to activate FIX in buffer was evaluated. Strikingly, PKa, but not FXIIa or plasmin, promoted FIXa formation in a concentration-dependent manner (Figure 4A), consistent with some previous reports,27,28 although this finding is controversial.29 Furthermore, PKa initiated TG in a dose-dependent manner when spiked into recalcified NPP (Figure 4B) or FXII-deficient (Figure 4C) or FXI-deficient (Figure 4D) plasma, but not FIX-deficient (Figure 4E) plasma. This profile in plasma was similar to that observed with RBC-MVs. PKa-induced TG was completely abolished in NPP in the presence of soybean trypsin inhibitor (SBTI), an inhibitor of PKa (Figure 4B). Because PKa was purified from human plasma, we analyzed it for purity (and specifically for possible FXIa contamination) using tandem mass spectrometry. Results showed that PKa represented ∼98.7% of total protein in the preparation. FXI was detected in an extremely low concentration (0.02% total protein), ruling out the possibility that FIX activation was caused by contaminating FXIa.
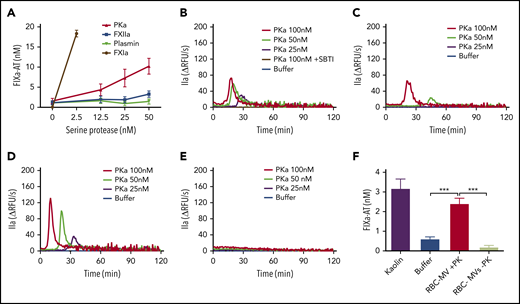
Procoagulant activity of purified human kallikrein in a purified system and in plasma. (A) FIXa-AT complexes generated after incubation of 100 nM purified human FIX with increasing concentrations of purified human kallikrein (red), FXIIa (blue), or plasmin (green) for 1 hour at 37°C, then addition of 2.5 µM purified human AT and 1 U/mL unfractionated heparin. Human FXIa (1.2 nM) was used as positive control (brown). TG in normal pooled (B), XII-deficient (C), XI-deficient (D), or IX-deficient (E) plasma spiked with increasing concentrations of purified human PKa. (F) FIXa-AT complexes formed after incubation for 150 minutes at 37°C of 100 nM purified human FIX with RBC-MVs in the presence (red) or absence (green) of 580 nM PK and 670 nM HK. Kaolin was used as positive control (purple bar). Buffer was used as negative control (B-F; blue). FIXa-AT was measured using an in-house ELISA capturing FIX and detecting AT. Data are expressed as means ± SD of at least 3 independent experiment run in duplicate. **P < .05; ***P < .001.
Procoagulant activity of purified human kallikrein in a purified system and in plasma. (A) FIXa-AT complexes generated after incubation of 100 nM purified human FIX with increasing concentrations of purified human kallikrein (red), FXIIa (blue), or plasmin (green) for 1 hour at 37°C, then addition of 2.5 µM purified human AT and 1 U/mL unfractionated heparin. Human FXIa (1.2 nM) was used as positive control (brown). TG in normal pooled (B), XII-deficient (C), XI-deficient (D), or IX-deficient (E) plasma spiked with increasing concentrations of purified human PKa. (F) FIXa-AT complexes formed after incubation for 150 minutes at 37°C of 100 nM purified human FIX with RBC-MVs in the presence (red) or absence (green) of 580 nM PK and 670 nM HK. Kaolin was used as positive control (purple bar). Buffer was used as negative control (B-F; blue). FIXa-AT was measured using an in-house ELISA capturing FIX and detecting AT. Data are expressed as means ± SD of at least 3 independent experiment run in duplicate. **P < .05; ***P < .001.
RBC-MVs activate FIX in the presence of plasma PK
We next assessed whether RBC-MVs can indirectly activate FIX via PK activation in buffer. Kaolin, RBC-MVs, or buffer were incubated with FIX in the presence of PK and its cofactor, HK. FIX activation was detected by measuring FIXa-AT complexes. Kaolin and RBC-MVs promoted the formation of levels of FIXa-AT complexes that were (respectively) five- and fourfold greater than the buffer control (Figure 4F). Levels of FIXa-AT were not greater than the buffer control when RBC-MVs were incubated with FIX and HK in the absence of PK (Figure 4F), indicating the dependence on PK activation. Furthermore, FXI was undetectable on RBC-MVs, by western blot (supplemental Figure 4), ruling out the possibility of direct activation of FIX by FXIa on RBC-MVs. These results strongly support the PKa-FIX pathway as an alternative to the canonical intrinsic pathway for coagulation activation induced by RBC-MVs.
Simultaneous inhibition of FXIIa and kallikrein abolishes TG initiated by RBC-MVs in plasma
Because RBC-MVs activated PK and PKa activated FIX, we hypothesized that the PKa-FIX pathway could be the alternate pathway by which RBC-MVs initiate TG in plasma, independent of FXII and FXI. Thus, we added CTI or SBTI alone or in combination with NPP to block FXIIa or PKa, respectively. CTI abolished kaolin-initiated TG, whereas SBTI had little effect, indicating that the PKa-FIX pathway does not play a major role in TG initiated by kaolin (Figure 5A).
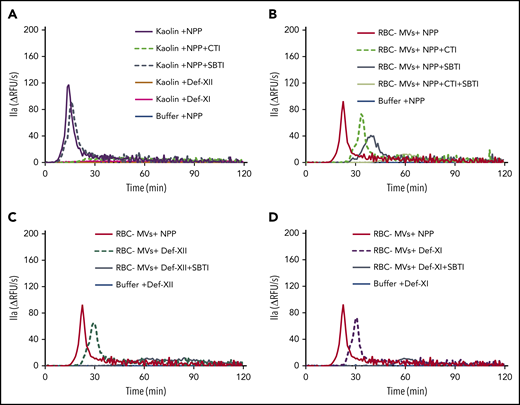
Effect of CTI and SBTI on TG initiated by RBC-MVs in plasma. (A) Effect of CTI (dashed green) and SBTI (hashed gray) on kaolin-initiated (30 µg/mL) TG in MV-free NPP. (B) Effect of CTI (hashed green) and SBTI (gray) on TG initiated by RBC-MVs in NPP. (C) Effect of SBTI (gray) on TG initiated by RBC-MVs in FXII-deficient, MV-free plasma, as compared with XII-deficient plasma alone (hashed dark green). (D) Effect of SBTI (gray) on TG initiated by RBC-MVs in FXI-deficient, MV-free plasma as compared with XI-deficient plasma alone (dashed purple). Data representative of 3 independent experiments. Buffer (blue) was used as the negative control (A-D), and NPP (red) was used as a comparator (B-D). Data are representative of 3 independent experiments.
Effect of CTI and SBTI on TG initiated by RBC-MVs in plasma. (A) Effect of CTI (dashed green) and SBTI (hashed gray) on kaolin-initiated (30 µg/mL) TG in MV-free NPP. (B) Effect of CTI (hashed green) and SBTI (gray) on TG initiated by RBC-MVs in NPP. (C) Effect of SBTI (gray) on TG initiated by RBC-MVs in FXII-deficient, MV-free plasma, as compared with XII-deficient plasma alone (hashed dark green). (D) Effect of SBTI (gray) on TG initiated by RBC-MVs in FXI-deficient, MV-free plasma as compared with XI-deficient plasma alone (dashed purple). Data representative of 3 independent experiments. Buffer (blue) was used as the negative control (A-D), and NPP (red) was used as a comparator (B-D). Data are representative of 3 independent experiments.
The pattern of thrombin inhibition was different when TG was initiated by RBC-MVs when compared with kaolin. CTI alone delayed and attenuated TG induced by RBC-MVs in MV-free NPP (Figure 5B) in a manner similar to FXII (Figure 5C) or FXI (Figure 5D) deficiency. These results are consistent with a partial contribution of the canonical (FXII/FXI-dependent) intrinsic pathway to TG induced by RBC-MVs in plasma. SBTI alone also delayed and attenuated RBC-MV–induced TG in NPP (Figure 5B). However, RBC-MV–induced TG was completely abolished in the presence of both CTI- and SBTI in NPP (Figure 5B), confirming that TG is dependent on both FXIIa and PKa generation. Interestingly, SBTI alone abolished TG induced by RBC-MVs in FXII-deficient (Figure 5C) or FXI-deficient (Figure 5D) plasma, indicating that the PKa-FIX pathway by itself can lead to robust TG. In aggregate, these data strongly support the existence of 2 independent pathways for initiation of TG in plasma after contact activation by RBC-MVs.
A protein factor plays a critical role in the initiation of RBC-MV–induced TG
We next sought to address the molecular mechanism by which RBC-MVs activate FXII and PK. Several biological compounds have been shown to activate the contact system, including inorganic polyphosphates, nucleic acids, PS, glycosaminoglycans, sulfatides, and misfolded proteins.30 As shown earlier, PS-containing synthetic lipid vesicles did not trigger TG in plasma. Treatment of RBC-MVs with polyphosphatases (supplemental Figure 5A-B) or glycosaminoglycan-degrading enzymes (supplemental Figure 5C) did not affect their ability to trigger TG in plasma. In addition, incubation of RBC-MVs with inhibitors of serine proteases, cysteine proteases, or calpain inhibitors did not significantly affect their TG capacity (supplemental Figure 5D-F). However, heating RBC-MVs at 60°C for 15 minutes inhibited their ability to activate FXII (Figure 6A) and PK (Figure 6B). As expected, heated RBC-MVs did not directly activate FXI (Figure 6C). When RBC-MVs were heated and resuspended in recalcified NPP, TG was abolished (Figure 6D; supplemental Figure 7B). Although addition of kaolin or tissue factor to NPP in the absence of synthetic lipid vesicles induce minimal thrombin formation, the addition of these vesicles restores robust TG (supplemental Figure 6A-B), indicating their ability to propagate TG because of their surface expression of anionic phospholipids. As expected, heating synthetic lipid vesicles at 60°C for 15 minutes did not affect this ability to propagate kaolin- or tissue factor–initiated TG (supplemental Figure 6A-B). Similarly, heating RBC-MVs did not interfere with their prothrombinase activity (supplemental Figure7A), nor did it result in any significant change in particle size or concentration (supplemental Figure 7C-D). Consistently, heated RBC-MVs added to kaolin or tissue factor–activated plasma restored robust TG (Figure 6D). These findings indicate that RBC-MVs possess the dual ability to both (1) initiate TG, mediated by a heat-sensitive factor; and (2) propagate TG that is likely mediated by prothrombinase activity. Supporting the possibility that the MV-associated activator is a protein, incubation of stored RBC-MVs with trypsin abrogated their capacity to initiate TG (Figure 6E), an effect that was not observed with trypsin-treated kaolin (Figure 6F). In aggregate, these experiments suggest that an unknown surface protein(s) on RBC-MVs initiates TG.
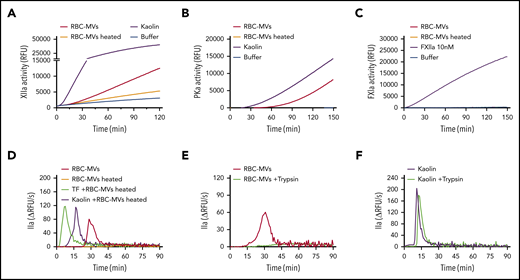
Effect of heating or incubation with trypsin on TG initiated by storage lesion–induced RBC-MVs. (A) RBC-MVs were heated (brown) or not (red) at 60°C for 15 minutes, then brought to room temperature before resuspension in a buffer system containing purified FXII and assayed for substrate cleavage. (B) RBC-MVs, heated (brown) or not (red), were resuspended in a buffer system containing purified PK and HK and assayed for substrate cleavage. Kaolin (purple) was used as the positive control (A-B). (C) Purified FXI was assayed for activation and substrate cleavage by RBC-MVs (red). Addition of FXIIa (purple) was used as the positive control, whereas sPL vesicles (green) were used as the negative control. Buffer (blue) was used as a control for residual active enzyme of the purified zymogens (A-C). (D) RBC-MVs, heated (brown) or not (red), were added to recalcified MV-free NPP for TG; 5 pM tissue factor (TF; green) or 30 µg/mL kaolin (purple) was used to initiate TG in recalcified NPP containing heated RBC-MVs. (E) RBC-MVs were incubated with buffer (red) or 20 µM bovine trypsin (green) overnight at 4°C followed by addition of 200 µM PPACK for 1 hour at room temperature to inhibit trypsin, before TG assay. (F) Kaolin (30 µg/mL, purple) and kaolin plus trypsin (green) were added to MV-free NPP supplemented with 4 µM synthetic lipid vesicles for TG.
Effect of heating or incubation with trypsin on TG initiated by storage lesion–induced RBC-MVs. (A) RBC-MVs were heated (brown) or not (red) at 60°C for 15 minutes, then brought to room temperature before resuspension in a buffer system containing purified FXII and assayed for substrate cleavage. (B) RBC-MVs, heated (brown) or not (red), were resuspended in a buffer system containing purified PK and HK and assayed for substrate cleavage. Kaolin (purple) was used as the positive control (A-B). (C) Purified FXI was assayed for activation and substrate cleavage by RBC-MVs (red). Addition of FXIIa (purple) was used as the positive control, whereas sPL vesicles (green) were used as the negative control. Buffer (blue) was used as a control for residual active enzyme of the purified zymogens (A-C). (D) RBC-MVs, heated (brown) or not (red), were added to recalcified MV-free NPP for TG; 5 pM tissue factor (TF; green) or 30 µg/mL kaolin (purple) was used to initiate TG in recalcified NPP containing heated RBC-MVs. (E) RBC-MVs were incubated with buffer (red) or 20 µM bovine trypsin (green) overnight at 4°C followed by addition of 200 µM PPACK for 1 hour at room temperature to inhibit trypsin, before TG assay. (F) Kaolin (30 µg/mL, purple) and kaolin plus trypsin (green) were added to MV-free NPP supplemented with 4 µM synthetic lipid vesicles for TG.
Discussion
We showed that RBC-MVs are progressively released into the supernatant of stored RBC concentrates over time. Most of the RBC-MVs had a diameter smaller than 200 nm and demonstrated PS-dependent procoagulant activity, in agreement with previous studies.18,31,32 These structural and functional properties appeared not to change over time in storage. Although RBCs do not express tissue factor, it was important to rule out tissue factor–initiated coagulation, as circulating tissue factor–bearing MVs may bind to other cells.33 Instead, we demonstrated that RBC-MVs initiate coagulation by direct and independent activation of both FXII and PK (Figure 7). In addition to well-recognized reciprocal activation between these 2 enzymes, this study adds that their active forms activate FIX via distinct pathways: (1) the canonical pathway in which FIX is activated by FXIa after FXI activation by FXIIa; and (2) a noncanonical pathway in which PKa directly activates FIX. To our knowledge, this is the first evidence that cell-derived MVs activate coagulation in plasma through a direct PKa-FIX pathway, although others previously described a similar pathway triggered by long-chain polyphosphates.27 However, we showed that treatment of RBC-MVs with polyphosphatases did not affect the ability to induce TG, and others failed to detect polyphosphates in stored RBC-MVs.16
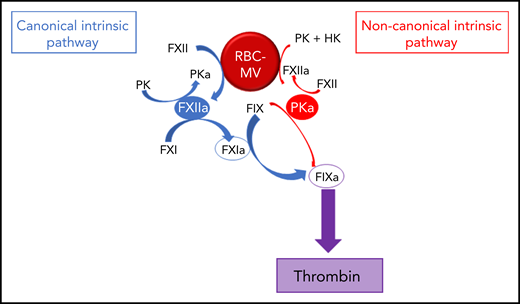
Pathways of initiation of coagulation by storage lesion–induced RBC-MVs. RBC-MVs directly promote FXII and PK activation, leading to FXIIa and PKa formation, respectively. By reciprocal activation, FXIIa further activates PK, whereas PKa further activates FXII. In addition, formation of these 2 enzymes leads to FIX activation via 2 independent pathways. The canonical pathway (in blue) in which FXIIa indirectly activates FIX after FXI activation, and an alternative pathway (in red) in which PKa directly activates FIX. Both pathways ultimately lead to thrombin formation via the common pathway.
Pathways of initiation of coagulation by storage lesion–induced RBC-MVs. RBC-MVs directly promote FXII and PK activation, leading to FXIIa and PKa formation, respectively. By reciprocal activation, FXIIa further activates PK, whereas PKa further activates FXII. In addition, formation of these 2 enzymes leads to FIX activation via 2 independent pathways. The canonical pathway (in blue) in which FXIIa indirectly activates FIX after FXI activation, and an alternative pathway (in red) in which PKa directly activates FIX. Both pathways ultimately lead to thrombin formation via the common pathway.
FXII binds directly,34,35 whereas PK binds indirectly to PS via HK.36 Whether this binding leads to proenzyme activation is controversial.34,35 In purified systems, we demonstrated that synthetic lipid vesicles failed to activate FXII, PK, FXI, or FIX. In a plasma milieu, these vesicles propagated, but did not initiate, TG. In contrast, we observed a dose-dependent TG when RBC-MVs were added to recalcified NPP supplemented with synthetic lipid vesicles, indicating that RBC-MVs were able to initiate TG in plasma, in addition to their well-known, PS-dependent, ability to propagate coagulation. More interesting, exposure of RBC-MVs to heat abolished their ability to initiate but not to propagate TG in plasma, indicating that initiation of TG requires a heat-sensitive factor distinct from procoagulant phospholipid. Because treatment of RBC-MVs by trypsin also abrogated TG, it is likely that surface proteins on these MVs plays a critical role in the initiation of coagulation by activating FXII or PK. However, the putative proteins are unlikely to be calpain or serine, cysteine, or metalloproteases, as incubation of RBC-MVs with inhibitors of these enzymes did not mitigate TG. Rojkjaer and Schmaier26 demonstrated FXII-independent activation of PK on endothelial surfaces mediated by a membrane-bound prolylcarboxypeptidase that was inhibited by CTI or phenylmethylsulfonyl fluoride.37 However, it is unlikely that prolylcarboxypeptidase is responsible for RBC-MV–induced coagulation activation, because neither CTI nor phenylmethylsulfonyl fluoride abolished RBC-MV–induced TG. The molecular mechanism by which RBC-MVs activate FXII and PK remains unknown, but is under active investigation in our laboratory.
In contrast to storage lesion–induced RBC-MVs, in another study, researchers did not observe TG triggered in FXII-deficient plasma by MVs shed from RBCs in response to calcium ionophore.7 These 2 populations of RBC-MVs exhibit major differences in their protein content and lipid raft proteins,38 which may influence their respective procoagulant activities. Moreover, it appears that the dominance of the canonical vs the PKa-FIX pathway during initiation of coagulation depends on the activator. For instance, we observed that although kaolin activated both FXII and PK in purified systems, genetic deficiency or inhibition of FXII or FXI abolished kaolin-induced TG, whereas inhibition of PKa using SBTI had only a modest effect. These data indicate a preference for the canonical pathway. In contrast, long-chain polyphosphates27 or oversulfated chondroitin sulfates39 mediate FXIIa-dependent PKa formation without activating FXI. In our study, selective inhibition of FXIIa or PKa had comparable effects on TG induced by storage lesion–induced RBC-MVs. The molecular mechanism determining the choice of the canonical vs the PKa-FIX pathway requires further investigation. In addition, it is challenging to quantify the relative contributions of the canonical vs the PKa-FIX pathways to TG induced by RBC-MVs Sun and Gailani40 reported that FXIa is at least 30 times more efficient than PKa as an ex vivo activator of FIX. Accordingly, the PKa-FIX pathway may be a secondary pathway for contact system–initiated TG if FXI is activated.
Although previous studies agree that FXII is not absolutely required for coagulation activation by storage lesion–induced RBC-MVs,8,16 the role of FXI is more controversial. We observed that TG is attenuated when storage lesion–induced RBC-MVs are resuspended in FXI-deficient plasma or in NPP in the presence of inhibitory anti-FXI antibodies, consistent with other reports.19 Likewise, anti-FXI antibodies induced a 50% attenuation of TG mediated by RBC-MVs isolated from the plasma of patients with sickle cell disease.41 In contrast, another study reported that TG was abolished in FXI-deficient plasma or in NPP in the presence of inhibitory anti-FXI antibodies.16 The reasons for these conflicting results are unclear. However, the difference between patient-derived and immunodepleted FXI-deficient plasma and the specificity of anti-FXI antibodies could be potential contributors to the discrepant results. Because of the similar structure and up to 58% homology in the amino acid sequence of FXI and PK, some anti-FXI antibodies may have cross-reactivity with PKa, leading to blockade of both the canonical and the noncanonical pathways. Indeed, the anti-FXI antibody used in this study inhibited PKa-induced TG in FXI-deficient plasma (data not shown). Likewise, immunodepletion of FXI in plasma could result in some depletion of PK. In addition, contact system activation during immunodepletion may further consume PK, potentially explaining the absence of TG induced by RBC-MVs in FXI-deficient plasma prepared by this approach.
The clinical significance of our findings remains to be demonstrated. Thrombin-AT complexes were more elevated in the plasma and the bronchoalveolar lavage fluid of patients undergoing cardiac surgery who underwent blood transfusion compared with those who did not,42 suggesting that RBC transfusion activates coagulation in vivo. Although stored RBCs may participate in the procoagulant and prothrombotic effects of blood transfusion through multiple mechanisms,43,44 existing evidence points to a significant role for RBC-MVs. Most of the prothrombinase activity observed in RBC concentrates is mediated by RBC-MVs.18 Infusion of such RBC-MVs into mice accelerated clot formation time, increased the α angle in thromboelastography, lowered the plasma fibrinogen level, and shortened the postinfusion tail bleeding time.45 In humans, it is challenging to determine the number of MVs that enter the circulation during RBC transfusion, because patients typically receive a variable number of RBC units that have been stored for variable durations. However, our data do suggest that the number of RBC-MVs entering the circulation increases with the number and storage duration of RBC concentrates. Transfusion of limited units of RBC concentrates, which are rapidly diluted in the circulation, may not introduce sufficient numbers of RBC-MVs to overcome physiologic anticoagulants and thereby trigger coagulation. However, patients receiving large volume transfusion may receive a sufficient quantity of RBC-MVs over a short period that may exceed the threshold for coagulation activation. In support of this, Dhillon et al13 reported a 25% incidence of venous thromboembolism in trauma patients receiving massive blood transfusion, with the number of transfused red cell units being the only variable associated with an increased risk for thrombosis. Spinella and colleagues46 reported that longer duration of RBC storage (>28 days) was also associated with an increased risk of deep vein thrombosis and mortality in trauma victims receiving more than 5 units of RBCs. Finally, a recent secondary analysis of the Pragmatic, Randomized Optimal Platelet, and Plasma Ratios (PROPPR; clinicaltrials.gov #NCT01545232) trial focused on outcomes in trauma patients receiving massive RBC transfusion (≥10 units). The number of packed RBCs ≥22 days old was independently associated with an increase in death within 24 hours.47 Thus, we suggest that future clinical studies of large-volume transfusion, particularly in trauma victims, consider including ancillary analyses to evaluate the number and procoagulant activity associated with transfused RBC units.
In a broader sense, whether blood transfusion routinely leads to contact activation in vivo also has not been established. However, hypotensive transfusion reactions occur despite prestorage leukoreduction,48 likely because of excess bradykinin formation resulting from PKa-mediated cleavage of HK. We postulate that RBC-MVs that promote PK activation also represent potential triggers for posttransfusion generation of kinins, which may play a role in some inflammatory reactions to RBC transfusion. More important, blockade of the contact pathway activation may represent a novel strategy to prevent thrombosis and perhaps other inflammatory complications of RBC transfusion. Indeed, genetic or pharmacological inhibition of contact system proteins protects against thrombosis in animals.49-53 Although a role for FXII in human thrombosis is unclear,54 coagulation factors of the intrinsic pathway have been attractive targets for potential new antithrombotic therapies,55,56 because of the minimal risk for bleeding. Interestingly, lanadelumab, a PKa inhibitor licensed for the treatment of hereditary angioedema also attenuates coagulation activation in treated patients.57
We acknowledge several limitations in this study. SBTI was used to inhibit PKa in plasma. SBTI is also a weak inhibitor of other serine proteases of coagulation including thrombin and FXIa.58,59 Although we used a concentration of SBTI at which we observed inhibition of PKa with minimal effect on TG initiated by tissue factor (data not shown) or kaolin, the results of this study should be replicated using more specific PKa inhibitors.
Low percentages of MVs expressing surface markers of platelets, leukocytes, or endothelial cells may be found in RBC concentrate units.32,60 It has also been reported that RBC-MVs with a diameter ≤200 nm are CD63+, whereas those >200 nm are not, indicating that the smaller entities are exosomes whereas the larger ones are membrane-derived MVs. Because methods used to analyze RBC-MVs in this study do not provide information regarding cellular origin, we cannot rule out the presence of a small number of MVs derived from nonerythroid cells. Thus, we cannot determine the relative contribution of exosomes vs MVs to the procoagulant activities described.
The establishment of contact system activation in vivo and its contribution to adverse transfusion reactions in humans could open the door to novel strategies to prevent transfusion reactions by blocking the activation of the intrinsic coagulation pathway. Such an approach would potentially inhibit thrombosis and inflammation without increasing the risk of bleeding.
Original data may be obtained by e-mail request to the corresponding author.
The online version of this article contains a data supplement.
The publication costs of this article were defrayed in part by page charge payment. Therefore, and solely to indicate this fact, this article is hereby marked “advertisement” in accordance with 18 USC section 1734.
Acknowledgments
The authors thank Wolfram Ruf for kindly providing the 5G9 anti-human tissue factor antibody, Jian Liu for the glycosaminoglycan-degrading enzymes, Victoria Madden (The Microscopy Services Laboratory, UNC Department of Pathology and Laboratory Medicine) for transmission electron microscopy analysis, and Olesia Gololobova (UNC Center for Nanotechnology in Drug Delivery) for the NTA and dynamic light-scattering analyses.
This study was funded by the National Institutes of Health (NIH) National Heart, Lung, and Blood Institute (grants UO1HL117659 [D.N. and N.S.K.] and T32HL007149-42 [P.E. and S.C.S.]). The Microscopy Services Laboratory (UNC Department of Pathology and Laboratory Medicine) is supported in part by an NIH, National Cancer Institute Cancer Center Core Support Grant (P30CA016086) to the UNC Lineberger Comprehensive Cancer Center.
Authorship
Contribution: D.F.N., M.M., M.W.H., A.I., P.E., T.R., R.P., D.M.M., and N.S.K. designed the experiments; I.W. and M.H. provided the red blood cell concentrate units; D.F.N., M.W.H., A.I., P.E., M.P., and S.C.S. performed the experiments; D.F.N. wrote the initial draft of the manuscript; N.S.K. oversaw all experiments and data interpretation; and all authors contributed to several revisions of the manuscript.
Conflict-of-interest disclosure: The authors declare no competing financial interests.
The current affiliation for D.F.N. is Department of Pathology, University of Alabama at Birmingham, Birmingham, AL.
Correspondence: Nigel S. Key, 8008B Mary Ellen Jones Building, CB #7035, 116 Manning Dr, Chapel Hill, NC 27599; e-mail: nigel_key@med.unc.edu.

This feature is available to Subscribers Only
Sign In or Create an Account Close Modal