

Key Points
Myeloid-specific integrin α9 modulates arterial thrombosis by enhancing NETosis.

Abstract
Evidence suggests that neutrophils contribute to thrombosis via several mechanisms, including neutrophil extracellular traps (NETs) formation. Integrin α9β1 is highly expressed on neutrophils when compared with monocytes. It undergoes affinity upregulation on neutrophil activation, and stabilizes adhesion to the activated endothelium. The role of integrin α9 in arterial thrombosis remains unexplored. We generated novel myeloid cell-specific integrin α9−/− mice (α9fl/flLysMCre+) to study the role of integrin α9 in arterial thrombosis. α9fl/fl littermates were used as controls. We report that α9fl/flLysMCre+ mice were less susceptible to arterial thrombosis in ferric chloride (FeCl3) and laser injury-induced thrombosis models with unaltered hemostasis. Neutrophil elastase-positive cells were significantly reduced in α9fl/flLysMCre+ mice concomitant with reduction in neutrophil count, myeloperoxidase levels, and red blood cells in the FeCl3 injury-induced carotid thrombus. The percentage of cells releasing NETs was significantly reduced in α9fl/flLysMCre+ mouse neutrophils stimulated with thrombin-activated platelets. Furthermore, we found a significant decrease in neutrophil-mediated platelet aggregation and cathepsin-G secretion in α9fl/flLysMCre+ mice. Transfusion of α9fl/fl neutrophils in α9fl/flLysMCre+ mice restored thrombosis similar to α9fl/fl mice. Treatment of wild-type mice with anti-integrin α9 antibody inhibited arterial thrombosis. This study identifies the potential role of integrin α9 in modulating arterial thrombosis.
Introduction
In addition to being the earliest cells to arrive at the site of vascular injury, leukocytes (predominantly neutrophils) influence multiple aspects of thrombosis by facilitating a coordinated interaction with endothelial cells and platelets.1 Depletion of neutrophils has been shown to inhibit thrombosis, which suggests their role in modulating platelet adhesion, activation, and coagulation.2,3 NETosis is characterized by the release of neutrophil extracellular traps (NETs), which are composed of chromatin and antimicrobial proteins (histones, myeloperoxidase, elastase, and cathepsin G). Activated platelets are known to promote NETosis4 mediated by P-selectin and high-mobility group box 1.5,6 NETs also bind to plasma proteins such as fibrinogen, von Willebrand factor, and fibronectin, all known to contribute toward the progression of arterial thrombosis.7
Integrin α9 (which is absent in platelets; supplemental Figure 1, available on the Blood Web site) is highly expressed on neutrophils, in contrast to monocytes.8 On activation, neutrophils undergo affinity upregulation followed by transmigration to the plasma membrane. Although the role of integrin α9 in stabilizing neutrophil adhesion to the activated endothelium is known,8 its role in the context of arterial thrombosis remains unexplored. We sought to determine whether myeloid-cell integrin α9 regulates arterial thrombosis by modulating the release of NETs. To investigate this, we generated novel myeloid cell-specific integrin α9−/− (α9fl/flLysMCre+) mice and evaluated arterial thrombosis using FeCl3 and laser injury-induced thrombosis models. To explore the underlying mechanisms, we determined the levels of myeloperoxidase (MPO) and cathepsin G (markers of neutrophil activation), along with the extent of NET formation by neutrophils. The results of our study suggest that myeloid-specific integrin α9 modulates arterial thrombosis, most likely by promoting NETosis.
Methods
Because of the word limit, detailed methods are provided in the supplemental Data.
Mice
To generate myeloid-specific α9−/− mice, we crossed α9fl/fl mice with LysMCre+ mice (supplemental Figure 2). α9fl/fl littermates were used as controls.
FeCl3 and laser injury-induced carotid thrombosis
The FeCl3 (7.5%) injury-induced carotid artery thrombosis was assessed by intravital microscopy, as described previously.9,10 Laser injury-induced mesenteric artery thrombosis was evaluated using the Micropoint laser ablation system (Andor Technology), as described previously.10,11 Assays on both the models were performed by the investigator blinded to the genotypes.
Statistical analysis
Data represent mean ± SEM and are analyzed by Student t test and repeated measures ANOVA. Shapiro-Wilk test was used to check normality, and Bartlett's test was used to check equal variance. The results were considered significant at P < .05.
Results and discussion
Given the early lethality associated with the development of α9 global knockout mice,12 we generated a cohort of α9fl/flLysMCre+ and littermate control α9fl/fl mice on a C57BL/6J background (supplemental Figure 2). As expected, the expression of the integrin α9 receptor was nearly undetectable in the neutrophils isolated from the bone marrow of α9fl/flLysMCre+ mice (Figure 1A). Previously, it was shown that the global deletion of α9 in mice results in defective granulopoiesis.13 In contrast, complete blood counts were comparable in the α9fl/fl and α9fl/flLysMCre+ mice (supplemental Table 1), suggesting that myeloid-cell specific integrin α9-deficient mice could be used as a genetic model to define the role of integrin α9 in arterial thrombosis. Eight- to 10-week-old male α9fl/fl and α9fl/flLysMCre+ mice were subjected to 7.5% FeCl3 induced-carotid artery thrombosis. α9fl/flLysMCre+ mice exhibited smaller thrombi, prolonged occlusion time, and a significant decrease in the rate of thrombus growth (P < .05 vs α9fl/fl; Figure 1B). Despite being less susceptible to arterial thrombosis, the tail bleeding time was comparable between α9fl/fl and α9fl/flLysMCre+ mice, suggesting that myeloid cell-specific deficiency of integrin α9 does not alter hemostasis (Figure 1C). The time to complete occlusion was comparable between α9fl/fl and α9+/+LysMCre+ mice, thus ruling out the nonspecific effects of Cre-recombinase expression (supplemental Figure 3). The MPO levels (Figure 1D), neutrophil elastase-positive cells (Figure 1E), and neutrophil count (supplemental Figure 4) in the carotid artery thrombus of α9fl/flLysMCre+ mice were substantially decreased, suggesting fewer neutrophils getting recruited to the site of injury (P < .05 vs α9fl/fl). Transfusion of α9fl/fl neutrophils in α9fl/flLysMCre+ mice restored thrombosis similar to α9fl/fl mice (Figure 1F). Treatment of wild-type (C57BL/6J) mice with anti-α9 immunoglobulin blocking antibody (55A2C) inhibited arterial thrombosis (Figure 1G).
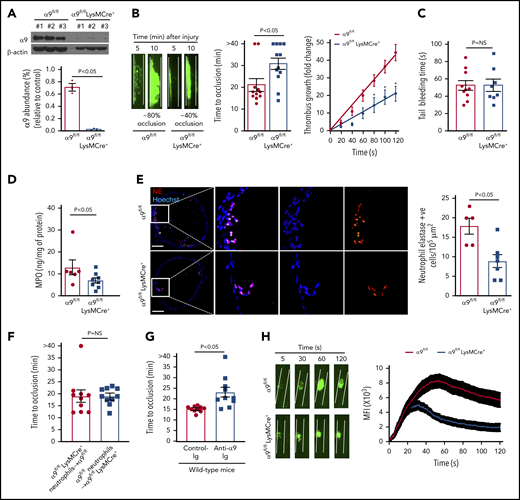
Myeloid specific α9−/− mice are less susceptible to experimental arterial thrombosis. (A) Western blot analysis of α9 integrin from bone marrow-derived neutrophils (top). Quantification (n = 3 mice/group) (bottom). (B) Representative microphotographs of carotid artery thrombus (7.5% FeCl3 injury) (left). Platelets were labeled ex vivo with calcein green. Time to occlusion (n = 11-12 mice/group) (middle). Rate of thrombus growth (n = 11-12 mice/group) (right). The rate of thrombus growth over a period of 2 minutes was calculated by dividing the area of the thrombus at time (n) by the area of the same thrombus at time (0) (defined as the time point at which the thrombus diameter first reached 30 μm). Slopes over time showed that the rate of thrombus growth in the α9fl/flLysMCre+ mice (slope, 0.1758) was decreased when compared with α9fl/fl mice (slope, 0.3575). *P < .05. (C) Tail bleeding assay (n = 8-10 mice/group). (D) MPO quantification by ELISA in carotid artery thrombus (n = 6-8 mice/group). (E) Representative immunofluorescence images of sections stained with anti-neutrophil elastase antibody. Scale bar, 100 μm. Boxed region is magnified to show colocalization of Hoechst/neutrophil elastase-positive cells (left). Quantification of neutrophil elastase-positive cells (n = 5-7 mice/group) (right). Value for each mouse represents a mean from 3 to 4 serial sections (each section separated by ∼70 μm). (F) Time to occlusion in α9fl/flLysMCre+ mice transfused with α9fl/fl mice neutrophils or α9fl/fl mice transfused with α9fl/fl LysMCre+ mice neutrophils (n = 10 vessels from 5 mice/group). (G) Time to occlusion in the wild-type mice (C57BL/6J) treated with anti-integrin α9 antibody or vehicle control (n = 10 vessels from 5 mice/group). (H) Representative microphotographs of mesenteric artery thrombus in the laser-injury model (left). Mean fluorescence intensity (MFI) over time (n = 19-22 vessels from 4 mice/genotype) (right).
Myeloid specific α9−/− mice are less susceptible to experimental arterial thrombosis. (A) Western blot analysis of α9 integrin from bone marrow-derived neutrophils (top). Quantification (n = 3 mice/group) (bottom). (B) Representative microphotographs of carotid artery thrombus (7.5% FeCl3 injury) (left). Platelets were labeled ex vivo with calcein green. Time to occlusion (n = 11-12 mice/group) (middle). Rate of thrombus growth (n = 11-12 mice/group) (right). The rate of thrombus growth over a period of 2 minutes was calculated by dividing the area of the thrombus at time (n) by the area of the same thrombus at time (0) (defined as the time point at which the thrombus diameter first reached 30 μm). Slopes over time showed that the rate of thrombus growth in the α9fl/flLysMCre+ mice (slope, 0.1758) was decreased when compared with α9fl/fl mice (slope, 0.3575). *P < .05. (C) Tail bleeding assay (n = 8-10 mice/group). (D) MPO quantification by ELISA in carotid artery thrombus (n = 6-8 mice/group). (E) Representative immunofluorescence images of sections stained with anti-neutrophil elastase antibody. Scale bar, 100 μm. Boxed region is magnified to show colocalization of Hoechst/neutrophil elastase-positive cells (left). Quantification of neutrophil elastase-positive cells (n = 5-7 mice/group) (right). Value for each mouse represents a mean from 3 to 4 serial sections (each section separated by ∼70 μm). (F) Time to occlusion in α9fl/flLysMCre+ mice transfused with α9fl/fl mice neutrophils or α9fl/fl mice transfused with α9fl/fl LysMCre+ mice neutrophils (n = 10 vessels from 5 mice/group). (G) Time to occlusion in the wild-type mice (C57BL/6J) treated with anti-integrin α9 antibody or vehicle control (n = 10 vessels from 5 mice/group). (H) Representative microphotographs of mesenteric artery thrombus in the laser-injury model (left). Mean fluorescence intensity (MFI) over time (n = 19-22 vessels from 4 mice/genotype) (right).
Next, to ensure that the observed effects are applicable to a broader context, susceptibility to arterial thrombosis was evaluated using another model: laser injury-induced mesenteric artery thrombosis. Consistent with FeCl3 injury-induced carotid artery thrombosis, α9fl/flLysMCre+ mice displayed significantly reduced thrombus growth (P < .05 vs α9fl/fl mice; Figure 1H). Susceptibility to FeCl3-induced carotid artery thrombosis was unaltered in monocyte-depleted (using clodronate liposomes) wild-type mice, demonstrating the minimal role of monocytes in the progression of arterial thrombosis in the conditions tested here (supplemental Figure 5). Together, these results suggest the potential role of integrin α9 expressed on neutrophils, but not on monocytes, toward the progression of arterial thrombosis.
It is known that N-formyl-methionyl-leucyl-phenylalanine (fMLP)–mediated stimulation and activation of neutrophils results in platelet aggregation.14 To further define the role of neutrophil integrin α9 on platelet activation, fMLP-stimulated neutrophil-induced platelet aggregation was determined in α9fl/fl and α9fl/flLysMCre+ mice. fMLP-activated neutrophils from α9fl/flLysMCre+ significantly impaired the onset and extent of platelet aggregation (P < .05 vs α9fl/fl mice; Figure 2A). Previous studies have suggested that the prothrombotic effects of neutrophils are significantly reduced on pharmacological inhibition or genetic deletion of cathepsin G (secreted from activated neutrophils and an important constituent of NETs).15,16 To determine whether cathepsin G release was affected by neutrophil integrin α9 deficiency, we measured and compared its level in the supernatant of fMLP-stimulated neutrophils from both α9fl/fl and α9fl/flLysMCre+ mice. Indeed, cathepsin G levels in the supernatant of activated neutrophils from α9fl/flLysMCre+ mice were found to be substantially reduced when compared with those of α9fl/fl mice (P < .05; Figure 2B). The underlying mechanism by which integrin α9 deficiency in neutrophils reduces the release of cathepsin G remains unclear and requires further investigation.
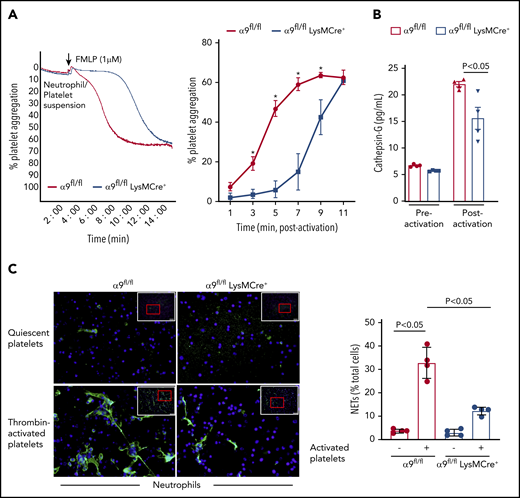
α9fl/fl LysMCre+ mice display reduced neutrophil mediated platelet aggregation and decreased NETosis. (A) Neutrophils from each genotype and wild-type platelets were stirred in a Chrono-log Whole Blood/Optical Lumi-Aggregometer (model 700-2) before the addition of fMLP. Representative aggregation curve for the fMLP stimulated neutrophils-induced platelet aggregation (left). Quantification (n = 4-5/group) (right). (B) Cathepsin G release as quantified by ELISA from the fMLP-stimulated neutrophils (n = 4 mice/group). (C) NETs assay was performed by stimulating neutrophils with thrombin-activated platelets. Representative microphotographs of NETs stained with PlaNET green (stains extracellular DNA, green) and counterstained with Hoechst (stains nuclei, blue) (left). Boxed region (lower magnification). Red insert in the boxed region is magnified and shown in the microphotographs. Quantification of the percentage of cells releasing NETs (n = 4 mice/group) (right). Value for each mouse represents a mean from 2 fields.
α9fl/fl LysMCre+ mice display reduced neutrophil mediated platelet aggregation and decreased NETosis. (A) Neutrophils from each genotype and wild-type platelets were stirred in a Chrono-log Whole Blood/Optical Lumi-Aggregometer (model 700-2) before the addition of fMLP. Representative aggregation curve for the fMLP stimulated neutrophils-induced platelet aggregation (left). Quantification (n = 4-5/group) (right). (B) Cathepsin G release as quantified by ELISA from the fMLP-stimulated neutrophils (n = 4 mice/group). (C) NETs assay was performed by stimulating neutrophils with thrombin-activated platelets. Representative microphotographs of NETs stained with PlaNET green (stains extracellular DNA, green) and counterstained with Hoechst (stains nuclei, blue) (left). Boxed region (lower magnification). Red insert in the boxed region is magnified and shown in the microphotographs. Quantification of the percentage of cells releasing NETs (n = 4 mice/group) (right). Value for each mouse represents a mean from 2 fields.
Next, we performed NET assay in vitro, using stimuli such as thrombin-activated platelets or TNF-α to determine whether integrin α9 modulates NETosis and, thereby, the progression of arterial thrombus. The percentage of cells releasing NETs was significantly reduced in α9fl/flLysMCre+ mice (P < .05 vs α9fl/fl mice; Figure 2C; supplemental Figure 6), suggesting that α9 may promote arterial thrombosis by enhancing NETosis. Previously, NETs were shown to promote red blood cell adhesion.7 Indeed, we found that RBC content (supplemental Figure 7) was decreased in the carotid thrombus of the α9fl/flLysMCre+ mice (P < .05 vs α9fl/fl). Collectively, a marked reduction in NETosis, MPO levels, neutrophil count, and red blood cells in the carotid artery thrombus concomitant with reduced neutrophil-mediated platelet aggregation, and cathepsin G levels from activated neutrophils in the α9fl/flLysMCre+/− mice provide potential mechanistic insights by which targeting integrin α9 may inhibit arterial thrombosis. In summary, these findings identify a novel role for integrin α9 in the modulation of arterial thrombosis.
For original data, please contact the corresponding author.
The online version of this article contains a data supplement.
The publication costs of this article were defrayed in part by page charge payment. Therefore, and solely to indicate this fact, this article is hereby marked “advertisement” in accordance with 18 USC section 1734.
Acknowledgments
The A.K.C. laboratory is supported by grants from the National Institutes of Health (NIH), National Heart, Lung, and Blood Institute (R35HL139926) and NIH, National Institute of Neurological Disorders and Stroke (R01NS109910), and by Established Investigator Award 18EIA33900009 from American Heart Association. N.D. is supported by a scholar award from the American Society of Hematology. M.K.N. and P.D. are supported by the American Heart Association postdoctoral award.
Authorship
Contribution: N.D. and M.K.N. performed experiments, analyzed results, and cowrote the manuscript; P.D. and M.J. performed experiments; G.D.F. performed experiments and edited the manuscript; S.K. provided the anti-integrin α9 antibody; and A.K.C. directed the project, interpreted results, and edited the manuscript.
Conflict-of-interest disclosure: The authors declare no competing financial interests.
Correspondence: Anil K. Chauhan, Department of Internal Medicine, University of Iowa, 3120 Medical Labs, Iowa City, IA 52242; e-mail: anil-chauhan@uiowa.edu.

This feature is available to Subscribers Only
Sign In or Create an Account Close Modal