

Key Points
MNT aids MYC-driven B lymphomagenesis by curbing MYC-induced apoptosis, primarily through suppressing BIM.
Induced MNT loss in transplanted Eμ-Myc lymphomas extends recipient survival, making MNT a novel therapeutic target for MYC-driven tumors.

Abstract
Deregulated overexpression of MYC is implicated in the development and malignant progression of most (∼70%) human tumors. MYC drives cell growth and proliferation, but also, at high levels, promotes apoptosis. Here, we report that the proliferative capacity of MYC-driven normal and neoplastic B lymphoid cells depends on MNT, a MYC-related transcriptional repressor. Our genetic data establish that MNT synergizes with MYC by suppressing MYC-driven apoptosis, and that it does so primarily by reducing the level of pro-apoptotic BIM. In Eμ-Myc mice, which model the MYC/IGH chromosome translocation in Burkitt’s lymphoma, homozygous Mnt deletion greatly reduced lymphoma incidence by enhancing apoptosis and markedly decreasing premalignant B lymphoid cell populations. Strikingly, by inducing Mnt deletion within transplanted fully malignant Eμ-Myc lymphoma cells, we significantly extended transplant recipient survival. The dependency of lymphomas on MNT for survival suggests that drugs inhibiting MNT could significantly boost therapy of MYC-driven tumors by enhancing intrinsic MYC-driven apoptosis.
Introduction
The transcription factor c-MYC (hereafter MYC) regulates expression of a multitude of genes involved in cell growth, proliferation, metabolism, and the DNA damage response.1 In normal cells, the level of MYC is tightly regulated, but in cancer cells, it is almost always elevated and constitutive.2,3 Although not fully transforming, MYC overexpression provides a strong drive toward malignancy.4 Importantly, however, MYC’s oncogenic potential is tempered by its propensity to induce apoptosis in cells stressed by inadequate access to cytokines or nutrients,5,6 particularly at high MYC levels.7 Mutations that inhibit apoptosis therefore synergize with MYC in tumorigenesis, as first shown for anti-apoptotic BCL-2.8,9
MYC and its closest relatives, N-MYC and L-MYC, bind DNA at canonical CACGTG E-boxes (and noncanonical variants) as a heterodimer with MAX, a related basic helix-loop-helix leucine zipper (bHLHLZ) protein.1 MYC:MAX heterodimers can activate10 or repress11 hundreds of genes,12 although many may be indirect targets.13 MYC action is opposed by other bHLHLZ relatives such as the 4 MXD proteins and MNT, which also heterodimerize with MAX and bind to E-boxes in many promoters and enhancers. By interacting with SIN3 proteins, MXD/MNT proteins recruit histone deacetylase-containing complexes to repress target genes.1
MNT14,15 is evolutionarily conserved and widely expressed during development and in adult tissues. Certain genes targeted by MYC:MAX heterodimers are also targets of MNT:MAX heterodimers.16 Genetic loss or knockdown of MNT was reported to enhance proliferation, increase RAS-induced transformation, and augment sensitivity to apoptotic stimuli, all characteristics of MYC overexpression.17-19 Therefore, MNT was posited as a tumor suppressor, a role supported by early mouse studies showing that its tissue-specific loss produced mammary adenocarcinomas20 and thymic lymphomas.21 Furthermore, MNT deletions have been noted in a variety of human cancers,22 including chronic lymphocytic leukemia23 and Sezary syndrome, a cutaneous T-cell lymphoma/leukemia.24
Surprisingly, however, recent studies indicate that MNT actually facilitates MYC-driven tumorigenesis, rather than acting as a tumor suppressor. Thus, Hurlin’s group found that T-cell-specific homozygous Mnt deletion prevented thymic lymphoma development in mice expressing a hypermorphic MYC protein (MYCT58A) in T cells,25 and we found that Mnt heterozygosity slowed T lymphomagenesis in VavP-Myc10hom mice and B lymphomagenesis in Eμ-Myc mice,26 which model the c-MYC/IGH chromosome translocations that hallmark Burkitt’s lymphomas.4 Link et al showed that MNT-null thymocytes expressing MYCT58A were more susceptible to apoptosis and proposed that MNT’s dominant physiological role is to suppress MYC-driven apoptosis.25 However, pre-B cells from Mnt+/− Eμ-Myc mice were not discernibly more sensitive to apoptosis than those from Mnt+/+ Eμ-Myc mice.26
To clarify the roles of MNT in B lymphopoiesis and lymphomagenesis, we have now undertaken conditional homozygous deletion of floxed Mnt alleles in wild-type (WT) and Eμ-Myc transgenic mice. In Eμ-Myc mice,4,27,28 constitutive Myc overexpression in B lymphoid cells, driven by the immunoglobulin H (IgH) enhancer Eμ, produces a polyclonal expansion of cycling nonmalignant pre-B cells, and every mouse goes on to develop a monoclonal malignant (transplantable) pre-B or B lymphoma harboring cooperating oncogenic mutations.29-31
We report here that B lymphopoiesis in both normal and Eμ-Myc mice is strikingly dependent on MNT. In its absence, B lymphoid cells are highly susceptible to apoptosis, largely as a result of upregulation of the BH3-only protein BIM, a potent pro-apoptotic BCL-2 family member.32,33 We show that Mnt deletion in Eμ-Myc mice greatly impedes B lymphomagenesis, and that inducing Mnt deletion in transplanted fully malignant Eμ-Myc lymphoma cells extends the survival of transplant recipients. These data provide genetic proof of principle that MNT would be an effective target for therapy of MYC-driven tumors.
Materials and methods
Mice
Mice used were Eμ-Myc,4,28 Bim−/−del339/1,34 Rag1Cre,35 Rosa26.CreERT2,36 and Mntfl/+,37 all on a C57BL/6 background. To activate CreER recombinase in Rosa26.CreERT2 mice, 5-week-old animals were given 200 mg/kg tamoxifen (T5648, Sigma-Aldrich) daily for 3 days by oral gavage, and analyzed after 4 weeks.
Nondecalcified long bone immunofluorescence
Immunofluorescence analysis was performed on long bone prepared without decalcification to maintain collagen signal, as described previously38,39 and in supplemental Methods, available on the Blood Web site. Quantification using ImageJ/FIJI “Analyze particle” function was performed blinded to genotype.
Statistical analysis
Statistical comparisons were made using unpaired 2-tailed Student’s t-test or analysis of variance with Prism v8.0 software (GraphPad, San Diego, CA), with P values ≤ .05 considered statistically significant. Mouse survival analysis was carried out using GraphPad Prism (Version 8.0), and significance determined using log-rank (Mantel-Cox) test.
Results
Mnt deletion largely prevents lymphomagenesis in Eμ-Myc mice
To investigate the physiological role of MNT in B lymphomagenesis and avoid the early embryonic lethality of homozygous Mnt deletion (∼E10 in C57BL/6 mice; K. J. Campbell, C.J.V., and S.C., unpublished results), we bred Eμ-Myc4 mice bearing floxed Mnt alleles (Mntfl/fl)37 and the Rag1Cre35 transgene, which expresses Cre recombinase only in lymphoid progenitor cells.
Lymphoid-specific Mnt deletion strikingly reduced lymphomagenesis in Eμ-Myc mice (Figure 1A). Mntfl/fl Eμ-Myc/Rag1Cre mice (blue curve) had a median survival of more than 400 days vs only 86 and 96 days, respectively, for Eμ-Myc (red) and Eμ-Myc/Rag1Cre (purple) mice. Consistent with our earlier study of Mnt+/− Eμ-Myc mice,26 mouse survival was also significantly extended in Eμ-Myc mice heterozygous for Mnt deletion (Mntfl/+ Eμ-Myc/Rag1Cre mice, lime green curve; median survival, 138 days).
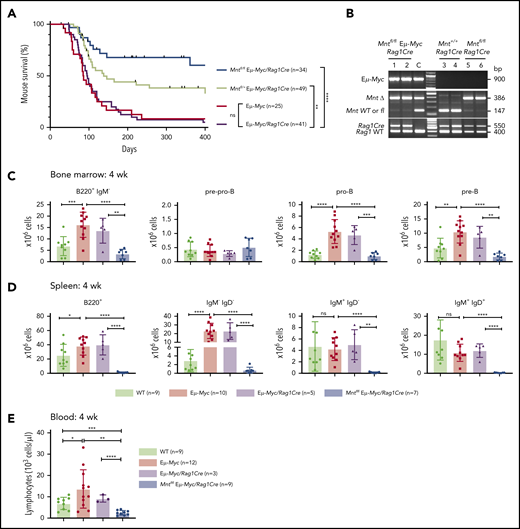
Lymphoid-specific loss of MNT greatly diminishes lymphoma development and induces lymphopenia in Eμ-Myc mice. (A) Kaplan-Meier survival curves showing reduced lymphoma development in Mntfl/fl Eμ-Myc/Rag1Cre mice (blue; median survival, 463 days) than Mntfl/+ Eμ-Myc/Rag1Cre mice (lime green; median survival, 138 days) and control Eμ-Myc mice (red; median survival, 86 days) and Eμ-Myc/Rag1Cre mice (purple; median survival, 96 days). **P ≤ .01; ****P ≤ .0001. Log-rank test. Killed mice showing no malignancy on autopsy were censored (black mark). X-axis was arbitrarily terminated at 400 days, but monitoring continued. (B) PCR analysis shows efficient deletion of floxed Mnt alleles by Rag1Cre in cells sorted from bone marrow of individual 4-week-old mice. WT and floxed Mnt alleles both produce 147-bp fragments, deleted Mnt allele (MntΔ), a 386-bp fragment. Lanes 1,2: pro-B and pre-B cells, respectively, from Mntfl/fl Eμ-Myc/Rag1Cre mouse #360; lanes 3,4: pre-B cells from control Mnt+/+Rag1Cre mice (#403, #404); lanes 5,6 pre-B cells from Mntfl/flRag1Cre mice (#413, #414); C, control DNA for Mnt PCRs. (C-D) Lymphoid-specific MNT loss induces lymphopenia. Flow cytometric quantification of B lymphoid subpopulations in bone marrow (C) and spleen (D) of 4-week-old WT (light green), Eμ-Myc (red), Eμ-Myc/Rag1Cre (purple), and Mntfl/fl Eμ-Myc/Rag1Cre (blue) mice. Supplemental Figure 3A exemplifies sorting strategy. Bar graphs show mean ± SD; *P ≤ .05; **P ≤ .01; ***P ≤ .001; ****P ≤ .0001. (E) Lymphocyte count in blood of 4-week-old mice of indicated genotypes, determined in an Advia hematology analyzer. Mean ± SD; *P ≤ .05; **P ≤ .01; ***P ≤ .001; ****P ≤ .0001.
Lymphoid-specific loss of MNT greatly diminishes lymphoma development and induces lymphopenia in Eμ-Myc mice. (A) Kaplan-Meier survival curves showing reduced lymphoma development in Mntfl/fl Eμ-Myc/Rag1Cre mice (blue; median survival, 463 days) than Mntfl/+ Eμ-Myc/Rag1Cre mice (lime green; median survival, 138 days) and control Eμ-Myc mice (red; median survival, 86 days) and Eμ-Myc/Rag1Cre mice (purple; median survival, 96 days). **P ≤ .01; ****P ≤ .0001. Log-rank test. Killed mice showing no malignancy on autopsy were censored (black mark). X-axis was arbitrarily terminated at 400 days, but monitoring continued. (B) PCR analysis shows efficient deletion of floxed Mnt alleles by Rag1Cre in cells sorted from bone marrow of individual 4-week-old mice. WT and floxed Mnt alleles both produce 147-bp fragments, deleted Mnt allele (MntΔ), a 386-bp fragment. Lanes 1,2: pro-B and pre-B cells, respectively, from Mntfl/fl Eμ-Myc/Rag1Cre mouse #360; lanes 3,4: pre-B cells from control Mnt+/+Rag1Cre mice (#403, #404); lanes 5,6 pre-B cells from Mntfl/flRag1Cre mice (#413, #414); C, control DNA for Mnt PCRs. (C-D) Lymphoid-specific MNT loss induces lymphopenia. Flow cytometric quantification of B lymphoid subpopulations in bone marrow (C) and spleen (D) of 4-week-old WT (light green), Eμ-Myc (red), Eμ-Myc/Rag1Cre (purple), and Mntfl/fl Eμ-Myc/Rag1Cre (blue) mice. Supplemental Figure 3A exemplifies sorting strategy. Bar graphs show mean ± SD; *P ≤ .05; **P ≤ .01; ***P ≤ .001; ****P ≤ .0001. (E) Lymphocyte count in blood of 4-week-old mice of indicated genotypes, determined in an Advia hematology analyzer. Mean ± SD; *P ≤ .05; **P ≤ .01; ***P ≤ .001; ****P ≤ .0001.
Most tumors that did arise in Mntfl/fl Eμ-Myc/Rag1Cre mice were pre-B or B lymphomas, which is typical for Eμ-Myc mice, although a few seemed more differentiated (supplemental Table 1). The tumor burden was comparable in sick Mntfl/fl Eμ-Myc/Rag1Cre mice and Mnt+/+ Eμ-Myc/Rag1Cre mice (supplemental Figure 1A-B) although the Mnt-deleted tumors were less frequently accompanied by leukemia (2/13 [15%] versus 25/34 [74%]; supplemental Figure 1B, panel 1). Most tumors analyzed (10/13, 77%) lacked detectable MNT at either the DNA or protein level, with the other 3 having escaped Mnt deletion by loss (#344) or aberrant activity (#611, #340) of the Rag1Cre transgene (supplemental Figure 1C; supplemental Table 1). Inactivation of the p19Arf/p53 pathway in MNT-null tumors (4/10, 40%) was similar to that reported for conventional Eμ-Myc lymphomas (supplemental Figure 1C),30,40 suggesting that selection against p53 is no higher in MNT-deficient than in MNT-proficient lymphomas.
Overall, these data indicate that MNT expression is highly advantageous for MYC-driven B lymphomagenesis.
Mnt deletion mediated by Rag1Cre provokes severe B lymphopenia in young Eμ-Myc mice
To investigate why MNT loss profoundly reduced lymphomagenesis in Eμ-Myc mice, we analyzed their premalignant phase. First, we compared expression of MNT and MYC protein in B lymphoid progenitor populations sorted from the bone marrow of young (4-week-old) WT and Eμ-Myc mice (supplemental Figure 2A). Transgenic MYC protein was low in pre-pro-B cells, but high in pro-B, pre-B, and recirculating IgM+ B cells, whereas endogenous MYC was below the detection limit. MNT was readily detectable in pre-pro-B cells from both WT and Eμ-Myc mice, and its level increased substantially with maturation. Quantification of intracellular MNT by flow cytometry indicated that MNT was 2.5- to 3-fold higher in Eμ-Myc pro-B, pre-B, and IgM+ cells than in their WT counterparts (supplemental Figure 2B).
Rag1Cre-mediated deletion of floxed Mnt was highly efficient, as shown by polymerase chain reaction (PCR) analysis of pro-B and pre-B cells (Figure 1B). The loss of MNT induced severe B lymphopenia in young Eμ-Myc mice. The several-fold increase in immature B lymphoid (B220+IgM−) cells normally seen in the bone marrow of young Eμ-Myc mice (red, purple bars)27 was nullified by the Mnt deletion in Mntfl/fl Eμ-Myc/Rag1Cre mice (blue bar; Figure 1C, panel 1; supplemental Figure 3A). Although pro-B and pre-B cells were both reduced ∼5-fold, the more immature pre-pro-B cell progenitors (in which Rag1-driven Cre expression is marginal) were unaffected (Figure 1C, panels 2-4). The MNT-null phenotype was even more extreme in the spleen (Figure 1D), where B lymphoid cells (B220+) were barely detectable, and the deficit affected all major subpopulations: pre-B (B220+IgM−IgD−), immature B (B220+IgM+IgD−) and mature follicular B (B220+IgM+IgD+). Circulating lymphocytes, normally elevated in young Eμ-Myc mice, were even lower in Mnt-deficient Eμ-Myc mice than in WT mice (Figure 1E).
Unsurprisingly, Mntfl/+ Eμ-Myc/Rag1Cre mice had a milder preleukemic phenotype than Mntfl/fl Eμ-Myc/Rag1Cre mice: B lymphoid cells were not significantly diminished in the bone marrow, but the spleen had a significant reduction (∼2-fold), and blood lymphocytes were also low (supplemental Figure 3B).
Mnt deletion mediated by CreERT2 also produces B lymphopenia
To confirm these results in an acute model of Mnt deficiency, we used the Rosa26.CreERT2 transgene (hereafter CreERT2), which encodes Cre recombinase fused to a modified hormone-binding domain of the estrogen receptor within the Rosa26 locus.36 Five-week-old Mntfl/fl Eμ-Myc/CreERT2 and control Eμ-Myc and Eμ-Myc/CreERT2 mice were treated on 3 successive days with tamoxifen by oral gavage and analyzed 4 weeks later (supplemental Figure 4A), to avoid early transient tamoxifen toxicity.41 PCR and immunoblot analyses of CD19+ splenocytes demonstrated highly efficient deletion of floxed Mnt alleles and barely detectable MNT protein in 5 independent experiments (eg, supplemental Figure 4B-C).
The bone marrow of tamoxifen-treated Mntfl/fl Eμ-Myc/CreERT2 mice had a significant deficit (2- to 3-fold) of B220+IgM− cells (supplemental Figure 4D). Although pre-pro-B cells were unaffected, both pro-B cells and pre-B cells were substantially reduced (∼3-fold) and splenic B lymphoid cells (B220+) were severely depleted (∼5-fold; supplemental Figure 4E).
Thus, MNT loss engineered by 2 independent approaches abrogated the MYC-driven lymphocyte expansion that normally hallmarks young Eμ-Myc mice. The reduced pool of proliferating premalignant early B lineage cells was undoubtedly a major contributing factor to the reduced lymphoma incidence in MNT-deficient Eμ-Myc mice.
MNT loss exacerbates Eμ-Myc-driven apoptosis
To identify the mechanism for the profound decrease in B lymphoid cells in young MNT-deficient Eμ-Myc mice, we analyzed MYC levels, cell proliferation, senescence, and apoptosis in pro-B and pre-B cells from the bone marrow of 4-week-old Mntfl/fl Eμ-Myc/Rag1Cre and control mice. MYC protein was substantially higher in Eμ-Myc pro-B and pre-B cells than in WT cells, as expected, but its level did not differ significantly between Eμ-Myc cells that had or lacked MNT (Figure 2A). Cell cycle analysis of bone marrow cells (Figure 2B) indicated that MNT loss did not diminish Eμ-Myc-driven cycling of pre-B cells,27 although pro-B cell cycling appeared marginally suppressed. The proportion of senescent cells was comparable in pro-B cells of all 3 genotypes, as measured by intracellular staining for H3K9 trimethylation,42 and although a small, nonsignificant increase was noted in pre-B cells expressing Eμ-Myc, MNT loss had no effect (Figure 2C).

Mnt deletion increases apoptosis but does not affect MYC protein level, cell cycling, or senescence in premalignant Eμ-Myc pro-B and pre-B cells. (A) MNT loss does not change MYC level. Pro-B and pre-B cells were sorted from bone marrow of 4-week-old mice, permeabilized and stained with MYC antibody (blue) or isotype-matched control (red). (Bottom) Mean intracellular MYC fluorescence (MFI) ± SD in WT (light green), Eμ-Myc (red), and Mntfl/fl Eμ-Myc/Rag1Cre (blue) pro-B and pre-B cells; n = 3 or 4; mean ± SD; **P ≤ .01; ns = not significant. (B) Cell cycle analysis. DNA content of 4′,6-diamidino-2-phenylindole-stained cells shows that the proportion of cycling (S-G2-M) pre-B cells in Mntfl/fl Eμ-Myc/Rag1Cre (blue) is comparable to that in Eμ-Myc (red) and Eμ-Myc/Rag1Cre (purple) mice. n = 3-6. Mean ± SD; *P ≤ .05; **P ≤ .01. (C) MNT loss does not significantly affect H3K9 trimethylation. Representative H3K9me3 staining of sorted permeabilized pro-B and pre-B cells from 4-week WT (light green), Eμ-Myc (red), and Mntfl/fl Eμ-Myc/Rag1Cre (blue) mice (left panels) and MFI for 3 mice of each genotype in 3 independent experiments (right panels). (D) MNT loss markedly elevates apoptosis. Annexin-V+ pro-B and pre-B cells were 2- to 3-fold more frequent in bone marrow of Eμ-Myc (red) than WT (light green) mice and ∼3-fold higher in Mntfl/fl Eμ-Myc/Rag1Cre (blue) than in Eμ-Myc mice. Top panels show typical fluorescence-activated cell sorting plots, and bottom panel shows mean percentage annexin-V+ cells; n = 6 to 9 individual mice; mean ± SD; **P ≤ .01; ***P ≤ .001.
Mnt deletion increases apoptosis but does not affect MYC protein level, cell cycling, or senescence in premalignant Eμ-Myc pro-B and pre-B cells. (A) MNT loss does not change MYC level. Pro-B and pre-B cells were sorted from bone marrow of 4-week-old mice, permeabilized and stained with MYC antibody (blue) or isotype-matched control (red). (Bottom) Mean intracellular MYC fluorescence (MFI) ± SD in WT (light green), Eμ-Myc (red), and Mntfl/fl Eμ-Myc/Rag1Cre (blue) pro-B and pre-B cells; n = 3 or 4; mean ± SD; **P ≤ .01; ns = not significant. (B) Cell cycle analysis. DNA content of 4′,6-diamidino-2-phenylindole-stained cells shows that the proportion of cycling (S-G2-M) pre-B cells in Mntfl/fl Eμ-Myc/Rag1Cre (blue) is comparable to that in Eμ-Myc (red) and Eμ-Myc/Rag1Cre (purple) mice. n = 3-6. Mean ± SD; *P ≤ .05; **P ≤ .01. (C) MNT loss does not significantly affect H3K9 trimethylation. Representative H3K9me3 staining of sorted permeabilized pro-B and pre-B cells from 4-week WT (light green), Eμ-Myc (red), and Mntfl/fl Eμ-Myc/Rag1Cre (blue) mice (left panels) and MFI for 3 mice of each genotype in 3 independent experiments (right panels). (D) MNT loss markedly elevates apoptosis. Annexin-V+ pro-B and pre-B cells were 2- to 3-fold more frequent in bone marrow of Eμ-Myc (red) than WT (light green) mice and ∼3-fold higher in Mntfl/fl Eμ-Myc/Rag1Cre (blue) than in Eμ-Myc mice. Top panels show typical fluorescence-activated cell sorting plots, and bottom panel shows mean percentage annexin-V+ cells; n = 6 to 9 individual mice; mean ± SD; **P ≤ .01; ***P ≤ .001.
Notably, however, MNT loss significantly exacerbated apoptosis of Eμ-Myc B lymphoid cells (Figure 2D). Annexin-V+ cells in bone marrow pro-B and pre-B cell populations of MNT-deficient Eμ-Myc mice (blue) were ∼3-fold higher than for regular Eμ-Myc mice (red), which in turn were ∼3-fold higher than for cells from WT mice (light green).
MNT loss increases apoptosis in situ in bone marrow
To complement these ex vivo analyses, we immunostained non-decalcified long bones38,39,43 from 4-week-old (premalignant) Eμ-Myc mice of all genotypes for CD19 (B lymphoid cells), Ki67 (proliferation), and cleaved caspase-3 (apoptosis) and collected a tilescan map by dual 2-photon/confocal microscopy. Representative stained images are presented in Figure 3A-B, and whole-bone staining is quantified in Figure 3C-E. The bones from Eμ-Myc and Eμ-Myc/Rag1Cre mice were packed with CD19+ and Ki67+ cells, as anticipated. In contrast, Mnt-deleted Eμ-Myc mice (Mntfl/fl Eμ-Myc/Rag1Cre) had far fewer CD19+ cells, consistent with analysis by flow cytometry (Figure 1C). The low level of Ki67-positive cells in bones isolated from Mntfl/fl Eμ-Myc/Rag1Cre mice undoubtedly reflects this B lymphopenia. Of note, cleaved caspase-3-positive cells were as prevalent in Mntfl/fl Eμ-Myc/Rag1Cre bones as in those from Eμ-Myc and Eμ-Myc/Rag1Cre mice (Figure 3E), consistent with a higher proportion of the B lymphoid cells being apoptotic.
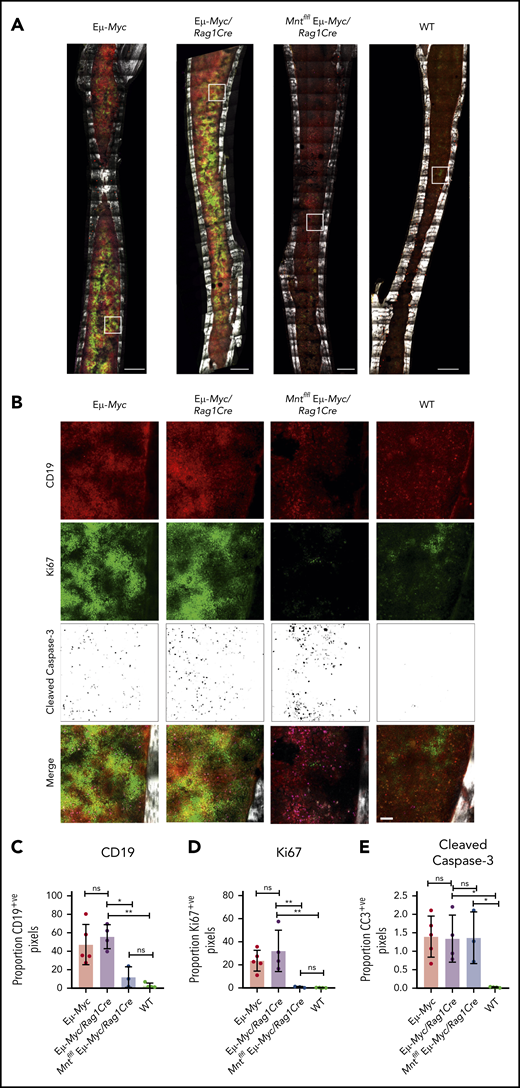
In situ analysis of cell proliferation and apoptosis. (A) Tilescan sections of whole tibia. Bones were harvested from 4-week old Eμ-Myc (n = 5), Eμ-Myc/Rag1Cre (n = 4), Mntfl/fl Eμ-Myc/Rag1Cre (n = 3), and WT (n = 4) mice, stained and imaged by combined 2-photon/confocal imaging of cell populations in situ. Red = CD19; green = Ki67; magenta = cleaved caspase-3; gray = second harmonic generation (bone). To better visualize CC3-positive cells in Figure 3B, the data are presented using an inverse black and white LUT (Lookup Table). Scale bar, 500 μm. (B) Zoomed areas from original tilescans in panel A (white boxes) with individual channels for CD19, Ki67, and cleaved caspase-3. A merged image (bottom panel) from each area is shown relative to bone signal (gray). Scale bar, 50 μm. (C-E) Quantification of cells in tibia positive for CD19, Ki67, and cleaved caspase-3. Color images were separated into single binary channels and then thresholded, and the number of positive pixels for each whole bone section quantified for CD19 (C), Ki67 (D) and cleaved caspase-3 (E). Each data point represents a whole quantified bone from an individual mouse in each genotype (n = 3-5). Bar graphs show mean ± SD; significance was determined by analysis of variance *P ≤ .05; **P ≤ .01).
In situ analysis of cell proliferation and apoptosis. (A) Tilescan sections of whole tibia. Bones were harvested from 4-week old Eμ-Myc (n = 5), Eμ-Myc/Rag1Cre (n = 4), Mntfl/fl Eμ-Myc/Rag1Cre (n = 3), and WT (n = 4) mice, stained and imaged by combined 2-photon/confocal imaging of cell populations in situ. Red = CD19; green = Ki67; magenta = cleaved caspase-3; gray = second harmonic generation (bone). To better visualize CC3-positive cells in Figure 3B, the data are presented using an inverse black and white LUT (Lookup Table). Scale bar, 500 μm. (B) Zoomed areas from original tilescans in panel A (white boxes) with individual channels for CD19, Ki67, and cleaved caspase-3. A merged image (bottom panel) from each area is shown relative to bone signal (gray). Scale bar, 50 μm. (C-E) Quantification of cells in tibia positive for CD19, Ki67, and cleaved caspase-3. Color images were separated into single binary channels and then thresholded, and the number of positive pixels for each whole bone section quantified for CD19 (C), Ki67 (D) and cleaved caspase-3 (E). Each data point represents a whole quantified bone from an individual mouse in each genotype (n = 3-5). Bar graphs show mean ± SD; significance was determined by analysis of variance *P ≤ .05; **P ≤ .01).
MNT loss also constrains B lymphoid development in WT mice
Because induction of apoptosis by MYC depends on its level of expression,7 whether MNT deficiency would affect normal B lymphopoiesis was unclear. To assess this, we compared B lymphoid cell subpopulations in 6-week-old WT, Rag1Cre, and Mntfl/flRag1Cre mice (exemplified in supplemental Figure 5). Notably, Mnt deletion significantly reduced B lymphoid cell numbers in both the bone marrow and spleen, although the reduction was less pronounced than in Eμ-Myc mice (Figure 4A-B). Tamoxifen-treated Mntfl/flCreERT2 mice also developed B lymphopenia, having fewer pre-pro-B, pro-B, pre-B, and B cells (Figure 4C-D). Thus, MNT loss impairs normal as well as Eμ-Myc-driven B lymphopoiesis.
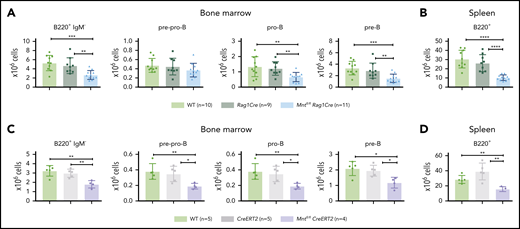
Loss of MNT impedes normal B lymphoid development. (A-B) Deletion of floxed Mnt by Rag1Cre provokes lymphopenia in mice lacking Eμ-Myc transgene. Quantification of B lymphoid subpopulations from bone marrow (A) and spleen (B) of 4-week-old WT (light green), Rag1Cre (green), and Mntfl/flRag1Cre (light blue) mice. Supplemental Figure 5A exemplifies sorting strategy. n indicates number of mice. Mean ± SD; **P ≤ .01; ***P ≤ .001; ****P ≤ .0001. (C-D) Activation of CreERT2 by tamoxifen also provokes B lymphopenia in mice lacking Eμ-Myc transgene. Quantification of indicated B lymphoid populations from bone marrow (C) and spleen (D). Young (6-week-old) WT (light green), CreERT2 (gray), and Mntfl/flCreERT2 (lavender) mice were treated for 3 days with tamoxifen by oral gavage and analyzed 4 weeks later. n indicates number of mice. Bar graphs show mean ± SD; *P ≤ .05; **P ≤ .01.
Loss of MNT impedes normal B lymphoid development. (A-B) Deletion of floxed Mnt by Rag1Cre provokes lymphopenia in mice lacking Eμ-Myc transgene. Quantification of B lymphoid subpopulations from bone marrow (A) and spleen (B) of 4-week-old WT (light green), Rag1Cre (green), and Mntfl/flRag1Cre (light blue) mice. Supplemental Figure 5A exemplifies sorting strategy. n indicates number of mice. Mean ± SD; **P ≤ .01; ***P ≤ .001; ****P ≤ .0001. (C-D) Activation of CreERT2 by tamoxifen also provokes B lymphopenia in mice lacking Eμ-Myc transgene. Quantification of indicated B lymphoid populations from bone marrow (C) and spleen (D). Young (6-week-old) WT (light green), CreERT2 (gray), and Mntfl/flCreERT2 (lavender) mice were treated for 3 days with tamoxifen by oral gavage and analyzed 4 weeks later. n indicates number of mice. Bar graphs show mean ± SD; *P ≤ .05; **P ≤ .01.
MNT constrains B lymphoid apoptosis largely by down-regulating expression of pro-apoptotic BIM
B lymphopoiesis in the bone marrow depends on signaling through the interleukin 7 (IL-7) receptor.44 Hence, to explore the mechanism underlying the enhanced apoptosis of MNT-deficient but otherwise normal B lymphoid cells, we cultured bone marrow-derived B progenitor cells (CD19+IgM−) in IL-7. In WT cells, endogenous MYC protein increased substantially by 48 hours, and MNT and anti-apoptotic MCL-1 also rose (Figure 5A). MNT deficiency had little effect on either the MYC level (supplemental Figure 6A) or the proliferation profile (supplemental Figure 6B) of pro-B cells cultured for 4 days in IL-7. Strikingly, however, apoptotic pro-B cells (annexin-V+) increased ∼3-fold (Figure 5B,E), despite the supraoptimal levels of IL-7.
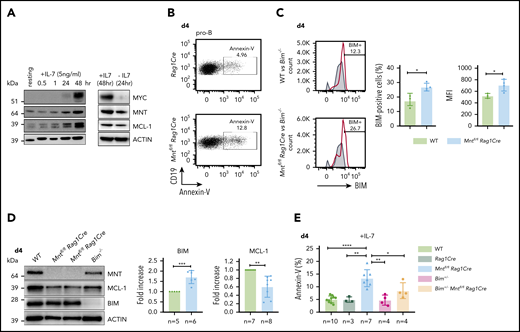
Loss of MNT increases apoptosis of pro-B cells in IL-7 cultures. (A) Expression of MYC, MNT, and MCL-1 protein in CD19+IgM− cells sorted by FACS from bone marrow of 6-week-old WT mice, before and during culture in IL-7, and 24 hours after IL-7 removal. (B-D) MNT-deficient pro-B cells exhibit increased apoptosis, elevated BIM, and decreased MCL-1. Pro-B cells were obtained by culturing CD19+ bone marrow cells (isolated using microbeads) in IL-7 (5 ng/ml) for 4 days. (B) Annexin-V staining of Rag1Cre and Mntfl/flRag1Cre pro-B cells; profiles are typical of those from ≥3 mice of each genotype (see panel E). (C) Flow cytometric analysis of intracellular BIM using Bim−/− cells (gray shaded) as a negative control. Bar graphs compare percentage BIM-positive cells and mean fluorescence intensity (MFI) from WT (light green) vs Mntfl/flRag1Cre (light blue) mice, determined in 3 independent experiments; MFI of BIM-null cells was subtracted from that of either WT or Mntfl/flRag1Cre cells stained in same experiment. (D) Typical western blot of pro-B cells from WT, Mntfl/flRag1Cre and control Bim−/− mice. For each blot, BIM and MCL-1 levels were quantified relative to ACTIN and normalized to WT value. Bar graphs show mean fold-change ± SD **P ≤ .01; ***P ≤ .001. (E) Bim heterozygosity significantly reduces apoptosis of MNT-null pro-B cells. Cells were stained with annexin-V after 4 days in IL-7. Bar graphs show mean ± SD; *P ≤ 0.05; **P ≤ .01; ****P ≤ .0001.
Loss of MNT increases apoptosis of pro-B cells in IL-7 cultures. (A) Expression of MYC, MNT, and MCL-1 protein in CD19+IgM− cells sorted by FACS from bone marrow of 6-week-old WT mice, before and during culture in IL-7, and 24 hours after IL-7 removal. (B-D) MNT-deficient pro-B cells exhibit increased apoptosis, elevated BIM, and decreased MCL-1. Pro-B cells were obtained by culturing CD19+ bone marrow cells (isolated using microbeads) in IL-7 (5 ng/ml) for 4 days. (B) Annexin-V staining of Rag1Cre and Mntfl/flRag1Cre pro-B cells; profiles are typical of those from ≥3 mice of each genotype (see panel E). (C) Flow cytometric analysis of intracellular BIM using Bim−/− cells (gray shaded) as a negative control. Bar graphs compare percentage BIM-positive cells and mean fluorescence intensity (MFI) from WT (light green) vs Mntfl/flRag1Cre (light blue) mice, determined in 3 independent experiments; MFI of BIM-null cells was subtracted from that of either WT or Mntfl/flRag1Cre cells stained in same experiment. (D) Typical western blot of pro-B cells from WT, Mntfl/flRag1Cre and control Bim−/− mice. For each blot, BIM and MCL-1 levels were quantified relative to ACTIN and normalized to WT value. Bar graphs show mean fold-change ± SD **P ≤ .01; ***P ≤ .001. (E) Bim heterozygosity significantly reduces apoptosis of MNT-null pro-B cells. Cells were stained with annexin-V after 4 days in IL-7. Bar graphs show mean ± SD; *P ≤ 0.05; **P ≤ .01; ****P ≤ .0001.
BIM, a potent BH3-only protein in the BCL-2 family,32,33 is a major apoptotic trigger during B lymphopoiesis.45-47 Flow cytometry and immunoblotting (Figure 5C-D) revealed higher BIM protein in cultured pro-B cells from Mntfl/flRag1Cre mice than in their WT counterparts, although Bim mRNA was unchanged (supplemental Figure 6D). MCL-1 protein (and RNA) levels fell in MNT-deficient pro-B cells (Figure 5D; supplemental Figure 6D), which would also increase susceptibility to apoptosis.
As BIM expression is haploinsufficient,31,45 we tested whether loss of a single Bim allele could ameliorate the pro-apoptotic effect of MNT loss. Indeed, the proportion of apoptotic pro-B cells in day 4 IL-7 cultures of Bim+/−Mntfl/fl Rag1Cre cells (light orange) was significantly lower than for Mntfl/fl Rag1Cre cells (light blue; Figure 5E). Bim heterozygosity did not, however, rescue the lower MCL-1 in IL-7 cultured pro-B cells lacking MNT (supplemental Figure 6C).
Taken together, these studies show that IL-7 receptor signaling upregulates MNT as well as MYC, and suggest that MNT reduces the MYC-induced apoptotic response, thereby facilitating MYC-driven cell production. They also suggest that the increased apoptosis of MNT-deficient pro-B cells is a result of elevated BIM levels, and probably also lower MCL-1.
Loss of a single Bim allele abrogates B lymphopenia provoked by MNT loss
These in vitro findings made it likely that the B lymphopenia induced in WT and Eμ-Myc mice by MNT loss reflected, at least in part, BIM-mediated apoptosis. Accordingly, MNT-deficient CD19+ spleen cells had more BIM protein than WT cells (compare lane 1 and 2 in Figure 6A), and BIM was even higher in splenocytes from Eμ-Myc mice (lane 4).

Bim heterozygosity rescues B lymphopoiesis, reduces apoptosis, and accelerates lymphomagenesis in MNT-deficient Eμ-Myc mice. (A) BIM and MCL-1 expression is elevated in MNT-deficient CD19+ cells. Cells were fluorescence-activated cell sorted from spleens of 4-week-old mice. (B-C) Bim heterozygosity largely restores B lymphopoiesis in young Mntfl/flRag1Cre mice. Flow cytometric enumeration of B lymphoid cells in bone marrow (B) and spleen (C) of 6-week-old mice. Data include mice in Figure 4 plus additional mice. Bar graphs show mean ± SD; *P ≤ .05; **P ≤ .01; ****P ≤ .0001. (D-E) Bim heterozygosity partially restores B lymphopoiesis in young MNT-deficient Eμ-Myc mice. Enumeration of B lymphoid cell populations in (D) bone marrow and (E) spleen of the indicated genotypes. Data for controls include certain mice in Figure 1C-D. Mean ± SD; *P ≤ .05; **P ≤ .01; ****P ≤ .001; ****P ≤ .0001. (F) Bim heterozygosity ameliorates enhanced apoptosis of pro-B and pre-B cells in the bone marrow of young MNT-deficient Eμ-Myc mice. Data include mice from Fig. 1C. Mean ± SD; *P ≤ .05; ns = not significant. (G) Bim heterozygosity accelerates lymphomagenesis in MNT-deficient Eμ-Myc mice. Kaplan-Meier survival curves showing enhanced morbidity of Bim+/−Mntfl/fl Eμ-Myc/Rag1Cre (orange) mice compared with Mntfl/fl Eμ-Myc/Rag1Cre (blue) mice, and of Bim+/−Mntfl/+ Eμ-Myc/Rag1Cre (mustard) mice compared with Mntfl/+ Eμ-Myc/Rag1Cre (lime green) mice. Survival curves for Eμ-Myc/Rag1Cre, Mntfl/+ Eμ-Myc/Rag1Cre, and Mntfl/fl Eμ-Myc/Rag1Cre mice are those shown in Figure 1A. Log-rank test; *P ≤ .05; ****P ≤ .0001.
Bim heterozygosity rescues B lymphopoiesis, reduces apoptosis, and accelerates lymphomagenesis in MNT-deficient Eμ-Myc mice. (A) BIM and MCL-1 expression is elevated in MNT-deficient CD19+ cells. Cells were fluorescence-activated cell sorted from spleens of 4-week-old mice. (B-C) Bim heterozygosity largely restores B lymphopoiesis in young Mntfl/flRag1Cre mice. Flow cytometric enumeration of B lymphoid cells in bone marrow (B) and spleen (C) of 6-week-old mice. Data include mice in Figure 4 plus additional mice. Bar graphs show mean ± SD; *P ≤ .05; **P ≤ .01; ****P ≤ .0001. (D-E) Bim heterozygosity partially restores B lymphopoiesis in young MNT-deficient Eμ-Myc mice. Enumeration of B lymphoid cell populations in (D) bone marrow and (E) spleen of the indicated genotypes. Data for controls include certain mice in Figure 1C-D. Mean ± SD; *P ≤ .05; **P ≤ .01; ****P ≤ .001; ****P ≤ .0001. (F) Bim heterozygosity ameliorates enhanced apoptosis of pro-B and pre-B cells in the bone marrow of young MNT-deficient Eμ-Myc mice. Data include mice from Fig. 1C. Mean ± SD; *P ≤ .05; ns = not significant. (G) Bim heterozygosity accelerates lymphomagenesis in MNT-deficient Eμ-Myc mice. Kaplan-Meier survival curves showing enhanced morbidity of Bim+/−Mntfl/fl Eμ-Myc/Rag1Cre (orange) mice compared with Mntfl/fl Eμ-Myc/Rag1Cre (blue) mice, and of Bim+/−Mntfl/+ Eμ-Myc/Rag1Cre (mustard) mice compared with Mntfl/+ Eμ-Myc/Rag1Cre (lime green) mice. Survival curves for Eμ-Myc/Rag1Cre, Mntfl/+ Eμ-Myc/Rag1Cre, and Mntfl/fl Eμ-Myc/Rag1Cre mice are those shown in Figure 1A. Log-rank test; *P ≤ .05; ****P ≤ .0001.
We therefore tested whether Bim heterozygosity would ameliorate B lymphopenia in MNT-deficient mice. Indeed, lowering BIM partially restored B220+ cell numbers in the bone marrow of young Mntfl/flRag1Cre mice (Figure 6B), and even more so in their spleen (Figure 6C) (compare light orange and light blue bars).
Bim heterozygosity also significantly ameliorated the B lymphopenia provoked by MNT loss in young Eμ-Myc mice. Total B lymphoid cells in the bone marrow (Figure 6D) and spleen (Figure 6E) of Bim+/− MNT-deficient Eμ-Myc mice were significantly more frequent than in Bim+/+ MNT-deficient Eμ-Myc mice (compare orange with blue bars), primarily as a result of more pro-B and pre-B cells. Annexin V staining of bone marrow pro-B and pre-B cells (Figure 6F) confirmed that Bim heterozygosity (orange) significantly reduced apoptosis of Mntfl/fl Eμ-Myc/Rag1Cre pro-B and pre-B cells (compare orange with blue bars).
Most strikingly, Figure 6G shows that the extended tumor-free survival afforded to Eμ-Myc mice by homozygous (blue) or even heterozygous Mnt (lime green) loss was greatly diminished by concomitant heterozygous loss of Bim (compare blue with orange curve and lime green with mustard curve). These observations raised the possibility that the MNT-deficient tumors arising at low frequency in Mntfl/fl Eμ-Myc/Rag1Cre mice might have suffered mutations or epigenetic changes that reduce BIM levels. Indeed, most of these tumors (10 of 13) did have markedly less BIM (supplemental Figure 1C; supplemental Table 1).
Eμ-Myc tumor cells remain dependent on MNT to curb MYC-induced apoptosis
Thus far, our results have shown that MNT plays a vital antiapoptotic role during early B lymphopoiesis, most strongly in Eμ-Myc, but also in WT, mice. We hypothesized, therefore, that the survival of fully malignant Eμ-Myc lymphoma cells might still require MNT. To test this, we exploited our Mntfl/fl Eμ-Myc/CreERT2 mice, which develop Eμ-Myc lymphomas carrying tamoxifen-deletable Mnt alleles (Figure 7A).

MNT loss induced in vivo in Eμ-Myc lymphomas extends survival of transplant recipients. (A) Schematic of experimental design. Mntfl/fl Eμ-Myc/CreERT2 and Eμ-Myc/CreERT2 mice were aged until they developed tumors (∼100 days). Tumor cells (Ly5.2+) were harvested and either cultured to establish cell lines for in vitro treatment with 4-OH tamoxifen (4-OHT) (see panel B) or injected intravenously into syngeneic nonirradiated C57BL/6-Ly5.1 mice for in vivo treatment with tamoxifen (see panel C). (B) 4-OH tamoxifen-induced Mnt loss enhances apoptosis of Eμ-Myc lymphoma cells in vitro. Short-term cell lines established from 2 independent Mntfl/fl Eµ-Myc/CreERT2 lymphomas (#1129 and #1271) and a control Eµ-Myc/CreERT2 lymphoma (#1194) were treated for 24 hours with or without 0.5 μM 4-OH tamoxifen and percentage annexin-V-positive cells determined by flow cytometry. Results are from 4 (#1194, #1129) or 3 (#1271) independent experiments; mean ± SD **P ≤ .01; **P ≤ .001. Immunoblot shows MNT and ACTIN protein in cells treated with tamoxifen or vehicle. (C) Tamoxifen-induced Mnt deletion significantly extends survival of mice transplanted with Mntfl/fl Eμ-Myc/CreERT2 lymphomas. Kaplan-Meier survival curves of mice transplanted with 14 independent Mntfl/fl Eμ-Myc/CreERT2 or 9 control Eμ-Myc/CreERT2 lymphomas and subsequently treated with either tamoxifen or vehicle alone. Each lymphoma was transplanted into 6 nonirradiated C57BL/6 recipients (2-4 × 106 cells/mouse), 3 of which were treated by oral gavage with tamoxifen and 3 with vehicle alone, for 2 successive days, starting on day 5; n indicates number of independent lymphomas transplanted. Significance was determined using log-rank test. See also Supplemental Figure 6A. (D) Induced Mnt deletion reduces viability of p53 wt and p53 mutant Mntfl/fl Eμ-Myc/CreERT2 lymphoma cell lines. Cell lines established from Mntfl/fl Eµ-Myc/CreERT2 and control Mnt+/+ Eµ-Myc/CreERT2 lymphomas were incubated with 0.5 μM 4-OH tamoxifen to delete Mnt, and cell viability was determined by flow cytometry (supplemental Figure 8). Viability of 4-OHT-treated cells at 48 and 72 hours is expressed relative to that of cells incubated in parallel without 4-OHT. P53 status, determined by treatment with nutlin3a, is indicated.
MNT loss induced in vivo in Eμ-Myc lymphomas extends survival of transplant recipients. (A) Schematic of experimental design. Mntfl/fl Eμ-Myc/CreERT2 and Eμ-Myc/CreERT2 mice were aged until they developed tumors (∼100 days). Tumor cells (Ly5.2+) were harvested and either cultured to establish cell lines for in vitro treatment with 4-OH tamoxifen (4-OHT) (see panel B) or injected intravenously into syngeneic nonirradiated C57BL/6-Ly5.1 mice for in vivo treatment with tamoxifen (see panel C). (B) 4-OH tamoxifen-induced Mnt loss enhances apoptosis of Eμ-Myc lymphoma cells in vitro. Short-term cell lines established from 2 independent Mntfl/fl Eµ-Myc/CreERT2 lymphomas (#1129 and #1271) and a control Eµ-Myc/CreERT2 lymphoma (#1194) were treated for 24 hours with or without 0.5 μM 4-OH tamoxifen and percentage annexin-V-positive cells determined by flow cytometry. Results are from 4 (#1194, #1129) or 3 (#1271) independent experiments; mean ± SD **P ≤ .01; **P ≤ .001. Immunoblot shows MNT and ACTIN protein in cells treated with tamoxifen or vehicle. (C) Tamoxifen-induced Mnt deletion significantly extends survival of mice transplanted with Mntfl/fl Eμ-Myc/CreERT2 lymphomas. Kaplan-Meier survival curves of mice transplanted with 14 independent Mntfl/fl Eμ-Myc/CreERT2 or 9 control Eμ-Myc/CreERT2 lymphomas and subsequently treated with either tamoxifen or vehicle alone. Each lymphoma was transplanted into 6 nonirradiated C57BL/6 recipients (2-4 × 106 cells/mouse), 3 of which were treated by oral gavage with tamoxifen and 3 with vehicle alone, for 2 successive days, starting on day 5; n indicates number of independent lymphomas transplanted. Significance was determined using log-rank test. See also Supplemental Figure 6A. (D) Induced Mnt deletion reduces viability of p53 wt and p53 mutant Mntfl/fl Eμ-Myc/CreERT2 lymphoma cell lines. Cell lines established from Mntfl/fl Eµ-Myc/CreERT2 and control Mnt+/+ Eµ-Myc/CreERT2 lymphomas were incubated with 0.5 μM 4-OH tamoxifen to delete Mnt, and cell viability was determined by flow cytometry (supplemental Figure 8). Viability of 4-OHT-treated cells at 48 and 72 hours is expressed relative to that of cells incubated in parallel without 4-OHT. P53 status, determined by treatment with nutlin3a, is indicated.
We first tested 2 short-term cell lines derived from independent primary Mntfl/fl Eμ-Myc/CreERT2 lymphomas. In both lines, exposure to 4-OH tamoxifen for 24 hours greatly reduced MNT levels, and annexin-V+ cells were ∼3-fold higher than in a control Mnt+/+ Eμ-Myc/CreERT2 line (Figure 7B). Thus, fully transformed Eμ-Myc-driven lymphoma cells depend on MNT for survival in vitro.
As a more stringent test, we induced Mnt deletion in vivo in transplanted primary lymphoma cells, predicting that MNT loss would disadvantage the tumor cells and hence prolong survival of transplant recipients. Fourteen independent Mntfl/fl Eμ-Myc/CreERT2 lymphomas and 9 independent Mnt+/+ Eμ-Myc/CreERT2 control lymphomas were transplanted into nonirradiated syngeneic mice (6 recipient mice per lymphoma), and once the lymphoma cells were established (5 days), 3 of the 6 recipients were treated with tamoxifen and 3 with vehicle alone. As expected, tamoxifen provided no survival benefit to recipients of control (Eμ-Myc/CreERT2) lymphoma cells (Figure 7C, lower panel). In marked contrast, after tamoxifen treatment, mice bearing Mntfl/fl Eμ-Myc/CreERT2 lymphoma cells survived significantly longer (Figure 7C, upper panel; P < .0001), whether transplanted with IgM+ or with IgM− tumors (supplemental Figure 7A). Comparable experiments with Mnt+/− Eμ-Myc/CreERT2 lymphomas showed that tamoxifen-induced loss of even a single Mnt allele significantly extended recipient survival (supplemental Figure 7B).
Strikingly, for 2 of the Mntfl/fl Eμ-Myc/CreERT2 tumors (#508 and #874), all (6/6) tamoxifen-treated transplant recipients survived for more than 19 weeks and were deemed “cured” (all their vehicle-treated recipients had required euthanasia within ∼30 days). Eleven other tumors had a median survival benefit of 8.3 days (range, 4-22 days), and 1 had none (supplemental Table 2). To address why the response rate varied, we analyzed CD19+ Ly5.2+ cells isolated by fluorescence-activated cell sorting from relapsing Mntfl/fl Eμ-Myc/CreERT2 lymphomas, comparing those treated with vehicle (as a surrogate for the primary tumor) and those treated with tamoxifen (supplemental Table 2; supplemental Figure 7C-D). For the nonresponding tumor (#676), PCR analysis indicated that failed Mnt deletion was a result of loss of the CreERT2 gene during tamoxifen treatment. The 11 other tumors analyzed retained readily detectable Mntfl DNA after treatment and still expressed significant MNT protein, suggesting they derived from cells that had escaped complete Mnt deletion. Thus, Figure 7C likely underestimates the true effect of MNT loss.
As 4/14 (29%) transplanted Mntfl/fl Eμ-Myc/CreERT2 lymphomas (including #508) lacked functional p53 (supplemental Figure 7D; supplemental Table 2), vulnerability to MNT loss did not appear to rely on WT p53. To directly test this, we established cell lines from independent primary Mntfl/fl Eμ-Myc/CreERT2 lymphomas and ascertained their p53 status by exposure to nutlin 3a48 (supplemental Figure 8). Figure 7D shows that 4/4 p53 WT Mntfl/fl Eμ-Myc/CreERT2 lines were extremely sensitive to apoptosis after Mnt deletion, and that 4/4 p53 mutant Mntfl/fl Eμ-Myc/CreERT2 lines were also vulnerable, albeit to a lesser degree.
Discussion
The proliferation of lymphocytes, similar to most cell types, requires MYC.49 MYC’s proliferative drive is checked, however, by its propensity to induce apoptosis at high expression levels.7 In this study, using conditional deletion in normal and Eμ-Myc mice, we provide strong genetic evidence that MYC antagonist MNT plays a vital role in both normal B lymphopoiesis and MYC-driven lymphomagenesis by reducing MYC-driven apoptosis. These results have important implications for the treatment of lymphomas and potentially other MYC-driven tumor types.
MNT was detectable at each major stage of B-cell development in the marrow of young WT mice, the highest levels being in pre-B and IgM+ B cells (supplemental Figure 2). Of note, MNT levels in Eμ-Myc mice were ∼3-fold higher than in comparable populations from WT mice.
Strikingly, homozygous deletion of floxed Mnt alleles in early B-cell progenitors (via Rag-1Cre) significantly extended the lifespan of Eμ-Myc mice by reducing the overall incidence of lymphomas (from >90% to ∼35%) and retarding their development (Figure 1A). The reduced lymphomagenesis in Mntfl/fl Eμ-Myc/Rag1Cre mice correlated with a profound early depletion of proliferating premalignant pro-B and pre-B cells (Figure 1C-E), the cells at risk of acquiring the oncogenic mutations needed for fully fledged malignancy.28 Enhanced apoptosis proved to be the major cause of this deficit, rather than reduced proliferation or increased cellular senescence (Figures 2 and 3). We conclude that MNT plays a major role in suppressing apoptosis driven by the high MYC levels in B lymphoid cells of Eμ-Myc mice.
Mnt deletion also reduced B lymphoid cell numbers in the bone marrow and spleen of normal mice (Figure 4), although the cell deficit was less than in Eμ-Myc mice. Thus, normal B-cell development also depends on MNT, perhaps because endogenous MYC levels can rise above the proapoptotic threshold at certain stages during their ontogeny. The most vulnerable population is likely to be the highly proliferative pro-B cell stage, in which MYC expression is highest (Figure 2A; see also the immgen.org database and Huang et al50 ), and which is dependent on IL-7 for proliferation and survival.51
These investigations of MNT function confirm and greatly extend previous genetic studies.25,26 Link et al reported that T lymphoid-specific Mnt deletion prevented thymic lymphoma development in transgenic mice expressing a mutant, more stable form of MYC, and that thymocytes lacking MNT were more susceptible to apoptosis.25 Our own previous study,26 using mice constitutively heterozygous for Mnt,37 found retarded lymphomagenesis in Eμ-Myc and VavP-MYC10 mice. However, there was no measurable enhancement of apoptosis in Mnt+/− Eμ-Myc B lymphoid cells. This disparity with our current findings undoubtedly reflects the greater effect of homozygous vs heterozygous loss of Mnt (compare Figure 1 and supplemental Figure 3B), but the germline deletion may also have selected for compensatory mechanisms to overcome developmental disadvantage.
In a major advance, we have established that the BH3-only protein BIM, a potent proapoptotic BCL-2 family member,32,33 is in part responsible for the increased apoptosis of MNT-deficient MYC-driven B lymphoid cells (Figure 6). BIM protein levels were higher in MNT-deficient pro-B cells (Figure 5C-D), apparently as a result of posttranscriptional mechanisms (supplemental Figure 6D), and tellingly, Bim heterozygosity reduced apoptosis of Mntfl/fl Eμ-Myc/Rag1Cre pro-B and pre-B cells (Figure 6F). Furthermore, lymphomas arose significantly faster and more frequently in Bim+/−Mntfl/fl Eμ-Myc/Rag1Cre than in Bim+/+Mntfl/fl Eμ-Myc/Rag1Cre mice (Figure 6G).
Although Bim heterozygosity substantially alleviated the B lymphopenia in premalignant MNT-deficient Eμ-Myc mice, it did not fully restore cell numbers (Figure 6 D-E). We speculate that lower MCL-1, as observed in cultured MNT-deficient pro-B cells (Figure 5D), may also have contributed to the enhanced apoptosis of MNT-deficient Eμ-Myc B lymphoid cells. Pertinently, MCL-1 is essential for the development and sustained growth of Eμ-Myc lymphoma cells.52,53
Another striking advance reported here is that even fully malignant (transplantable) Eμ-Myc-driven cells remain MNT-dependent (Figure 7). 4OH-tamoxifen-induction of Mnt-deletion in cell lines from Mntfl/fl Eμ-Myc/CreERT2 lymphomas rapidly induced cell death in vitro, particularly in lines retaining p53 functionality, but also in those lacking it (Figures 7B,D). Furthermore, when multiple Mntfl/fl Eμ-Myc/CreERT2 lymphomas were each transplanted into several immunocompetent syngeneic mice, recipients treated with tamoxifen to induce Mnt deletion in the tumor cells uniformly lived longer than those treated with vehicle alone (Figure 7C; P = .0002). Indeed, the true survival benefit of MNT loss is likely underestimated by this experiment because relapsing lymphoma cells lacked complete Mnt deletion. Conversely, the cures achieved for all recipients of 2 lymphomas likely reflected highly efficient Mnt loss.
This study clearly establishes that the proliferative capacity of MYC-driven B lymphocytes and lymphoma cells depends on MNT suppression of apoptosis. Thus, in this cellular setting, Mnt is a synergistic oncogene rather than a tumor suppressor, as earlier studies suggested,20,21,23,24 although MNT might well have a tumor suppressor role in certain cell types or circumstances.
Elevated MYC levels are common in lymphoid and many other malignancies,3 and often correlate with poor prognosis (eg, Barrans et al54 ). In several tumor models, MYC downregulation can elicit tumor regression (eg, Felsher and Bishop,55 , Meyer and Penn,56 and Soucek et al57 ). To date, however, drugs that directly target MYC have largely disappointed in the clinic, probably because reducing MYC leads to reduced MYC-driven apoptosis, and new approaches are urgently needed.58,59
Our genetic demonstration that MNT loss is a synthetic lethal vulnerability in MYC-driven lymphomas, even those mutant for p53 (Figure 7D), elevates MNT as an exciting prospect for drug development. We suggest that a therapeutic that inhibits or degrades MNT would serve as a Trojan horse by unleashing the intrinsic potential of MYC-driven lymphoma cells for self-destruction by apoptosis. Assessment of MNT and MYC protein levels in different human tumor types may reveal which malignancies might benefit from MNT inhibitors. A drug that substantially lowers MNT should not only elevate spontaneous apoptosis in MYC-driven tumors but also exacerbate sensitivity to diverse cytotoxic agents. Thus, co-targeting MNT could lift the efficacy of many current cytotoxic therapies, including the emerging BH3 mimetic drugs showing such high potential in blood cell cancers.33 Although a transcription factor such as MNT is a more challenging drug target than an enzyme, a growing number of epigenetic targets are showing promise,60 and targeted degradation has exciting potential.61
The online version of this article contains a data supplement.
The publication costs of this article were defrayed in part by page charge payment. Therefore, and solely to indicate this fact, this article is hereby marked “advertisement” in accordance with 18 USC section 1734.
Acknowledgments
The authors thank Peter Hurlin (Shriner’s Hospital for Children) for his kind gift of Mntfl/+ and Mnt+/− mice; our colleagues Jerry Adams, Andreas Strasser, Steve Nutt, Gemma Kelly, and Philippe Bouillet for comments on the manuscript and useful discussions; Andreas Strasser and Lorraine O’Reilly for fluorescent antibodies; Gemma Kelly, Marco Herold, and Kerstin Brinkmann for advice re tamoxifen activation protocol; Krystal Hughes, Crystal Stivala, Cassandra D'Alessandro, and Giovanni Siciliano for mouse husbandry; Simon Monard for assistance with flow cytometry; and Peter Maltezos for preparation of the figures.
This work was supported by grants from the National Health and Medical Research Council (program grant 461221) (S.C.) and the Leukemia & Lymphoma Society (Specialized Center of Research) grant 7001-13 (S.C.), National Health and Medical Research Council project grants GNT1060179 and GNT1122783 (A.P.N.), Leukemia and Lymphoma Society USA 6552-18, National Health and Medical Research Council APP1145442 and an RD Wright Fellowship APP1159488 (E.D.H.), an Australia Postgraduate Award (J.R.), philanthropic support to the Walter and Eliza Hall Institute, and operational infrastructure grants through the Australian Government Independent Research Institute Infrastructure Support Scheme and the Victorian Government Operational Infrastructure Support.
Authorship
Contribution: H.V.N., C.J.V., and M.R.R. performed most of the experiments; A.P.N. performed histological analysis; N.S.A. helped establish crosses; J.R. and E.D.H. performed and analyzed bone marrow imaging; and S.C. conceived the project, wrote the manuscript with input from authors, and secured funding.
Conflict-of-interest disclosure: The authors declare no competing financial interests.
The current affiliation for N.S.A. is Australian Centre for Blood Diseases, Monash University, Melbourne, VIC, Australia.
Correspondence: Suzanne Cory, The Walter and Eliza Hall Institute of Medical Research, 1G Royal Parade, Parkville, VIC 3052, Australia; e-mail: cory@wehi.edu.au.

This feature is available to Subscribers Only
Sign In or Create an Account Close Modal