

Key Points
Profiling of immune cell populations and plasma markers at day 100 post-HSCT demonstrates biological differences between cGVHD and L-aGVHD.
Immune profiling differences between patients meeting NIH diagnostic criteria and those with distinctive features only were similar.

Abstract
Human graft-versus-host disease (GVHD) biology beyond 3 months after hematopoietic stem cell transplantation (HSCT) is complex. The Applied Biomarker in Late Effects of Childhood Cancer study (ABLE/PBMTC1202, NCT02067832) evaluated the immune profiles in chronic GVHD (cGVHD) and late acute GVHD (L-aGVHD). Peripheral blood immune cell and plasma markers were analyzed at day 100 post-HSCT and correlated with GVHD diagnosed according to the National Institutes of Health consensus criteria (NIH-CC) for cGVHD. Of 302 children enrolled, 241 were evaluable as L-aGVHD, cGVHD, active L-aGVHD or cGVHD, and no cGVHD/L-aGVHD. Significant marker differences, adjusted for major clinical factors, were defined as meeting all 3 criteria: receiver-operating characteristic area under the curve ≥0.60, P ≤ .05, and effect ratio ≥1.3 or ≤0.75. Patients with only distinctive features but determined as cGVHD by the adjudication committee (non-NIH-CC) had immune profiles similar to NIH-CC. Both cGVHD and L-aGVHD had decreased transitional B cells and increased cytolytic natural killer (NK) cells. cGVHD had additional abnormalities, with increased activated T cells, naive helper T (Th) and cytotoxic T cells, loss of CD56bright regulatory NK cells, and increased ST2 and soluble CD13. Active L-aGVHD before day 114 had additional abnormalities in naive Th, naive regulatory T (Treg) cell populations, and cytokines, and active cGVHD had an increase in PD-1− and a decrease in PD-1+ memory Treg cells. Unsupervised analysis appeared to show a progression of immune abnormalities from no cGVHD/L-aGVHD to L-aGVHD, with the most complex pattern in cGVHD. Comprehensive immune profiling will allow us to better understand how to minimize L-aGVHD and cGVHD. Further confirmation in adult and pediatric cohorts is needed.
Introduction
An estimated 22 000 Americans and Canadians are long-term pediatric hematopoietic stem cell transplant (HSCT) survivors. In these survivors, the most serious nonrelapse complication is chronic graft-versus-host disease (cGVHD), a condition in which donor immune cells attack the recipient’s tissues as foreign. One in 4 (25%) pediatric and 60% of adult bone marrow transplant (BMT) survivors experience cGVHD, which causes long-term and often irreversible organ damage and a mortality rate at least twice as high compared with HSCT patients without cGVHD.1
Murine models of cGVHD have been imperfect and incompletely recapitulate human clinical cGVHD. Thus, human biomarker-based studies most accurately represent the biology of human cGVHD. Numerous human biomarker studies have focused on targeted aspects of cGVHD, including the role of T, B, regulatory T (Treg), regulatory B, and regulatory natural killer (NKreg) cells, as well as inflammatory cytokines.2-21 Very few studies have analyzed human cGVHD with a comprehensive approach to understand the interaction of a broader range of cellular immune populations and changes in plasma proteins. Using a comprehensive B-cell profile, we found concomitant alterations in adult cGVHD with higher CD19+CD10−CD27−CD21low B cells and lower T1 transitional CD27−CD10+ B cells.22 These studies support a comprehensive immune profiling approach using multiple cellular and plasma biologic variables to characterize patterns of cGVHD.
The National Institutes of Health consensus criteria (NIH-CC) identified late acute GVHD (aGVHD) as a clinical entity distinct from cGVHD presenting with symptoms of GVHD limited to involvement of the skin, liver, and gastrointestinal system with no sclerotic changes after day +100 posttransplant.23 While the biology of aGVHD in the first 3 months after transplant has been well studied, whether late aGVHD has biology similar to early aGVHD is unknown.
Recently, we concluded the ABLE study (Applied Biomarkers in Late Effects of Childhood Cancer), an international network of 27 pediatric BMT centers working in collaboration with the Pediatric Blood and Marrow Transplantation Consortium (PBMTC; designated ABLE/PBMTC 1202).24 ABLE/PBMTC1202 enrolled 302 children and included comprehensive central adjudication of the clinical grading of cGVHD and aGVHD and central immune phenotypic and cytokine analysis for each patient. We evaluated the immune phenotypic and plasma protein markers at 100 days after HSCT to evaluate for immune profiles that associate with the future development of late aGVHD and cGVHD. Of the 241 evaluable patients, 41 and 30 developed cGVHD and late aGVHD after day 114, respectively. There were 132 (controls) who never developed either late aGVHD or cGVHD. This group included patients with and without previous aGVHD that had resolved by day 100. An additional 28 cases were characterized as active (or onset) late aGVHD, and 10 others had active cGVHD. These patients had GVHD present either at day 100 or before day 114, allowing comparison of those who would develop cGVHD in the future (after day 114) vs patterns at the onset of disease (on or before day 114).
Methods
ABLE/PBMTC 1202 cGVHD biomarker study design
Twenty-seven pediatric transplant centers (6 Canadian, 20 United States, and 1 Austrian) enrolled 302 patients between August 2013 and February 2017 in accordance with the Declaration of Helsinki. Ethics committee approval was obtained at all participating institutions. The clinical study was recently described in detail.24 Patients were placed into 5 groups (see Figure 1): (1) a control group with no late aGVHD and no cGVHD (n = 132), (2) late aGVHD only (n = 30), (3) cGVHD (including overlap syndrome; n = 41), (4) active aGVHD day +100 to 114 (n = 28), or (5) active cGVHD before day +114 (n = 10). Centers graded patients according to the 2005 NIH-CC, with review by a study adjudication committee composed of experts in cGVHD. A patient met the NIH-CC for cGVHD by either having a distinctive feature plus a supporting laboratory test or having ≥1 diagnostic feature. An adjudication committee classified “non-NIH-CC” cGVHD cases as having a “distinctive” feature without a supporting laboratory test. Cases where aGVHD features were present concurrently with ≥1 diagnostic and/or distinctive cGVHD manifestation with supporting test were classified as overlap syndrome and included as a cGVHD case.
Shipping of samples
Two different types of tubes were used for sample collection: heparinized tubes for plasma (BD Vacutainer) and Cyto-chex BCT tubes (Streck; Canada distributor: Inter Medico, Markham, ON, Canada) for immunophenotyping. All peripheral blood samples were shipped to the Transplantation Applied Biomarkers laboratory at BC Children’s Hospital Research Institute in Vancouver, BC, Canada via FedEx overnight priority shipping (delivered within 24 hours after blood collection). For plasma isolation and storage, upon sample delivery, plasma was isolated from the blood cellular component by primary centrifugation. Plasma aliquots were kept frozen at −80°C until usage. The tubes were shipped at room temperature overnight and phenotyping performed on the same day of sample delivery.
Phenotyping procedure
Five panels were designed to look for different subpopulations in T, B, and NK cells. All antibodies, corresponding conjugated dyes, clones, and vendors, are provided in supplemental Table 7 (available at the Blood Web site). One hundred microliters of blood was used for all panels except for the Treg panel, where 200 μL blood was used. Samples were stained in the dark for 12 minutes at room temperature followed by treatment with fix/red blood cell lyze solution (eBiosceinces, Thermo Fisher Scientific, Waltham, MA). For intracellular staining, cells were made permeable using BD Perm II solution (BD Biosciences Mississauga, ON, Canada). Flow cytometry data were acquired using BD LSR Fortessa X-20 Special Order four channel flow cytometer (BD Biosciences, San Jose, CA). A minimum of 300 000 events were acquired for all panels. Instrument settings was also standardized using SPHERO Rainbow Calibration particles 6 peaks (Sphereotech, Lake Forest, IL) to adjust laser power drifts over time. Flow cytometry analysis files were analyzed using Kaluza software v2 (Beckman Coulter, Mississauga, ON, Canada). Flow cytometry accuracy and reproducibility were ensured by the approaches described in detail in supplemental Figure 1 with minimal batch variability.
Cytokine measurement
Batches of plasma samples were thawed, and 12 markers were analyzed, including ST2, osteopontin, soluble B-cell activating factor, soluble CD25 (sCD25), TIM-3, MMP3, ICAM-1, CXCL10, CXCL9, CXCL11, Reg3α, and soluble aminopeptidase N (sCD13). CXCL9 and CXCL11 were measured using an electrochemiluminescence dual-plex plate (Meso Scale Diagnostics, Gaithersburg, MD). sCD13 was measured using a colorimetric assay based on enzymatic activity, as previously described.4 The remaining cytokines were measured by standard colorimetric enzyme-linked immunosorbent assay (R&D Systems, Minneapolis, MN). We found a high accuracy, reproducibility, and linearity for all assays measuring soluble biomarkers and a high stability of analytes upon 24-hour shipment (supplemental Figure 2).
Statistical analysis
Univariate logistic regression was applied to contrast cGVHD (or late aGVHD) against controls for each marker, with the following clinical variables modeled as confounding factors: (1) prophylaxis or treatment with either alemtuzumab or antithymocyte globulin (ATG), (2) prophylaxis or treatment with rituximab, (3) recipient age, (4) the use of a peripheral blood donor product or not, and (5) whether the donor was HLA identical or not. Patients developing cGVHD before day 114 were included as active cGVHD, and those with aGVHD manifestations between days 100 and 114 were considered active late aGVHD. Patients were assigned to an “immune-tolerant” control group if they never developed early aGVHD, late aGVHD, or cGVHD at any time point after transplant. All patients with overlap syndrome were assigned to the cGVHD group. All 3 criteria were used to highlight biologically relevant markers: (1) P ≤ .05, (2) receiver-operating characteristic (ROC) area under the curve (AUC) ≥0.60, and (3) effect ratio of ≥1.3 or ≤0.75. The P value of each marker was estimated based on the Wald test. ROC AUC was computed by estimating the true-positive rate (proportion of cGVHD or late aGVHD correctly classified) against the false-positive rate (proportion of controls falsely classified as cGVHD or late aGVHD) for different marker thresholds. The effect ratio was calculated as the average marker value of patients with cGVHD (or late aGVHD) divided by the average marker value of controls. As our analyses were exploratory, no statistical adjustments were made for multiple testing. Given that there were numerous tests conducted, the probability of a type I error likely exceeded .05, but this was moderated by the additional ROC AUC and effect ratio criteria. For visualization of group differences, we applied t-distributed stochastic neighbor embedding (t-SNE)25 on the relevant markers to project their values onto a 2-dimensional subspace, where subjects with similar marker values would lie close to each other. This was performed in a completely unsupervised manner to reveal differences among cGVHD, aGVHD, and controls. To explicitly examine age-related differences and minimize sample size differences, each of the subject groups (cGVHD, aGVHD, and controls) were divided at their respective median age (∼12.5 years, ∼6.75 years, and 9 years) and analyzed for each age group (ie, younger cGVHD vs younger controls). To explicitly examine the effect of cGVHD onset time, we divided the cGVHD subjects into 2 onset groups, earlier (median onset ≤166 days) and later (median onset >166 days), compared with controls. All analyses were performed using MATLAB (MathWorks, Natwick, MA).
Results
No differences between cGVHD cases meeting NIH-CC and non-NIH-CC cGVHD cases
Previously, we found that ∼15% of pediatric cGVHD cases did not meet formal NIH-CC but had enough cGVHD features that the ABLE study committee agreed these patients most likely had cGVHD (called non-NIH-CC cases).24 Many of these patients received immune-suppressive therapy by the HSCT center identical to that applied to cGVHD, with clinical responses. After adjustment for major clinical factors, we defined a biologically significant marker only if it met all 3 of the following criteria: (1) a ROC AUC ≥0.60; (2) P ≤ .05, and (3) effect ratio ≥1.3 or ≤0.75. If they only met 1 or 2 of these criteria, then they were defined as not biologically significant. We used the following definitions of the cell populations as outlined in Table 1.8,26-37 We wished to determine whether there were biologic differences between these 2 groups by comparing the immune profile of the non-NIH-CC group (n = 6) with the NIH-CC group (n = 35). We found no significant differences between the 2 groups except for a decrease in the PD-1+ memory Th-cell population (PD-1+CD45RA−CD4+CD3+ T cells; P = .05; ROC AUC = 0.69; effect ratio = 0.66) in the non-NIH-CC group. Because there were no other major differences between the 2 populations, subsequent analyses included non-NIH-CC patients together with NIH-CC patients.
Phenotyping population definitions
| Cell population | Phenotype | References |
|---|---|---|
| B cells | ||
| CD19+ B cells | CD19+ lymphocytes | 26-29 |
| Nonswitched memory B cells | CD27+ IgD+ % of CD19+ B cells) | 26 |
| Naive B cells | CD38−CD10− B cells (% of CD19+ B cells) | 27 |
| Mature naive B cells | IgD+CD27−CD19+ | 26 |
| CD21low B cells | CD19+CD21low CD10+/− / CD19+ B cells | 28 |
| Transitional B cells | ||
| T2 transitional B cells | CD38intCD10int IgD+CD27− CD19+ B cells | 29 |
| T3 transitional B cells | CD38dim CD10low IgD+CD27−CD19+ B cells | 29 |
| T cells | ||
| T cells | CD3+T cells | |
| Tc cells | CD3+CD8+ T cells | |
| Th cells | CD3+CD4+ T cells | |
| Naive Th cells | CD31−CD45RA+CD4+CD3+ T cells | 30,31 |
| Naive Tc cells | CD31−CD45RA+CD8+CD3+ T cells | 30,31 |
| Activated T cells | CD3+CD69+ T cells | |
| CD31+ naive Th cells | CD31+CD45RA+CD4+CD3+ T cells | 30,31 |
| PD-1+ naive Tc cells | PD-1+CD45RA+ CD8+CD3+ T cells | 32 |
| Memory Th cells | PD-1-CD45RA−CD4+CD3+ T cells | 32 |
| Early Th | CD127+(IL-7Rα) CD25int CD4+CD3+ | 33 |
| Regulatory T cells | ||
| CD31+ naive Treg cells | CD31+CD45RA+CD127lowCD3+CD25+ T cells | 34 |
| Naive Treg cells | CD31−CD45RA+CD127lowCD3+CD25+ T cells | 34 |
| PD-1+ memory Treg | PD-1+CD45RA−CD127lowCD3+CD25+ T cells | 32 |
| PD-1− memory Treg | PD-1−CD45RA−CD127loCD3+CD25+ T cells | 27 |
| NK cells | ||
| CD56dim NK cells | CD56dim NK cells | 8 |
| Activated CD56dim NK cells | CD56dimCD69+ NK cells | 8 |
| Cytolytic CD56bright NK cells | CD56bright Ghi or CD335hiCD56brightPhi | 35-37 |
| NKregcells | ||
| NKreg cells | CD56brightCD335+ | 8 |
| NKreg noncytolytic | CD56brightCD3−Plow or CD56brightCD3−Glow | 35-37 |
| Cell population | Phenotype | References |
|---|---|---|
| B cells | ||
| CD19+ B cells | CD19+ lymphocytes | 26-29 |
| Nonswitched memory B cells | CD27+ IgD+ % of CD19+ B cells) | 26 |
| Naive B cells | CD38−CD10− B cells (% of CD19+ B cells) | 27 |
| Mature naive B cells | IgD+CD27−CD19+ | 26 |
| CD21low B cells | CD19+CD21low CD10+/− / CD19+ B cells | 28 |
| Transitional B cells | ||
| T2 transitional B cells | CD38intCD10int IgD+CD27− CD19+ B cells | 29 |
| T3 transitional B cells | CD38dim CD10low IgD+CD27−CD19+ B cells | 29 |
| T cells | ||
| T cells | CD3+T cells | |
| Tc cells | CD3+CD8+ T cells | |
| Th cells | CD3+CD4+ T cells | |
| Naive Th cells | CD31−CD45RA+CD4+CD3+ T cells | 30,31 |
| Naive Tc cells | CD31−CD45RA+CD8+CD3+ T cells | 30,31 |
| Activated T cells | CD3+CD69+ T cells | |
| CD31+ naive Th cells | CD31+CD45RA+CD4+CD3+ T cells | 30,31 |
| PD-1+ naive Tc cells | PD-1+CD45RA+ CD8+CD3+ T cells | 32 |
| Memory Th cells | PD-1-CD45RA−CD4+CD3+ T cells | 32 |
| Early Th | CD127+(IL-7Rα) CD25int CD4+CD3+ | 33 |
| Regulatory T cells | ||
| CD31+ naive Treg cells | CD31+CD45RA+CD127lowCD3+CD25+ T cells | 34 |
| Naive Treg cells | CD31−CD45RA+CD127lowCD3+CD25+ T cells | 34 |
| PD-1+ memory Treg | PD-1+CD45RA−CD127lowCD3+CD25+ T cells | 32 |
| PD-1− memory Treg | PD-1−CD45RA−CD127loCD3+CD25+ T cells | 27 |
| NK cells | ||
| CD56dim NK cells | CD56dim NK cells | 8 |
| Activated CD56dim NK cells | CD56dimCD69+ NK cells | 8 |
| Cytolytic CD56bright NK cells | CD56bright Ghi or CD335hiCD56brightPhi | 35-37 |
| NKregcells | ||
| NKreg cells | CD56brightCD335+ | 8 |
| NKreg noncytolytic | CD56brightCD3−Plow or CD56brightCD3−Glow | 35-37 |
G, granzyme; P, perforin.
Differences and similarities in day 100 immune populations and cytokines between cGVHD and late aGVHD
Changes in a number of B-cell populations were found at day 100 before the development of cGVHD (Figure 2A; supplemental Table 1), including overall B-cell counts, transitional T2 and T3 B cells, and CD21low B cells.11,12 We reported CD21low B cells either as a percentage of CD19+ B cells or as an absolute number of CD21low B cells, both of which were increased in our adult cGVHD cohort.22 Only the absolute CD21low B-cell value was significantly lower in the ABLE pediatric cohort (Figure 2A). Late aGVHD had a similar decrease in transitional B-cell pattern. Unlike cGVHD, late aGVHD had an increase in unswitched memory B cells (Figure 2B).
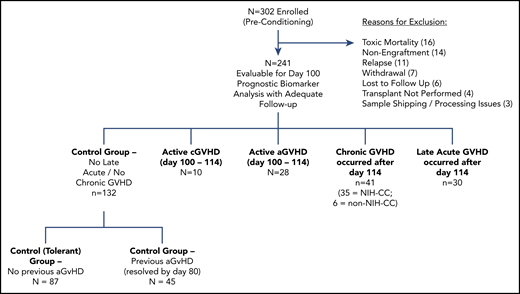
Evaluation of immune profiles at day 100 in patients who went on to develop late aGVHD or cGVHD. Volcano plots showing significant markers that meet all 3 criteria of a (1) P ≤ .05 (y-axis), (2) ROC AUC of ≥0.60 (circle, ≥0.60; cross, <0.6), and (3) effect ratio ≥1.3 or ≤0.75 (x-axis). The subfigures correspond to (A) cGVHD compared with no-cGVHD controls; (B) late aGVHD compared with no-cGVHD controls, and (C) cGVHD compared with late aGVHD. Cell population are identified by color, with dark blue representing B cells, orange myeloid populations, yellow NK cells, purple NKreg cells, green T cells, light blue Treg cells, and dark red plasma cytokines. We note the following clinical variables were modeled as confounding factors in the logistic regression model: (1) prophylaxis or treatment with either alemtuzumab or ATG, (2) prophylaxis or treatment with rituximab, (3) recipient age, (4) the use of a peripheral blood donor product or not, and (5) whether the donor was HLA identical or not.
Evaluation of immune profiles at day 100 in patients who went on to develop late aGVHD or cGVHD. Volcano plots showing significant markers that meet all 3 criteria of a (1) P ≤ .05 (y-axis), (2) ROC AUC of ≥0.60 (circle, ≥0.60; cross, <0.6), and (3) effect ratio ≥1.3 or ≤0.75 (x-axis). The subfigures correspond to (A) cGVHD compared with no-cGVHD controls; (B) late aGVHD compared with no-cGVHD controls, and (C) cGVHD compared with late aGVHD. Cell population are identified by color, with dark blue representing B cells, orange myeloid populations, yellow NK cells, purple NKreg cells, green T cells, light blue Treg cells, and dark red plasma cytokines. We note the following clinical variables were modeled as confounding factors in the logistic regression model: (1) prophylaxis or treatment with either alemtuzumab or ATG, (2) prophylaxis or treatment with rituximab, (3) recipient age, (4) the use of a peripheral blood donor product or not, and (5) whether the donor was HLA identical or not.
Unlike late aGVHD (Figure 2B), cGVHD was characterized by differences in T-cell populations compared with non-cGVHD controls (Figure 2A; supplemental Table 2). Children who developed cGVHD after day 114 had increases in the percentages of peripheral naive CD4+ (helper T [Th]; CD4+CD45RA+CD31−), naive PD-1+ CD8+ (cytotoxic T [Tc]; CD8+CD45RA+PD-1+) T cells, and activated T cells (CD3+CD69+) compared with non-cGVHD patients. By contrast, late aGVHD had no significant changes in these populations but did have a significant decrease peripheral Tc cells (Figure 2B; supplemental Table 2). Changes in classic activated cytolytic CD56dim NK cells were observed at day 100 in the cGVHD cohort (Figure 2A; supplemental Table 2) compared with late aGVHD that had an association in a cytolytic CD56bright NK population (granzyme B high; Figure 2B). NKT cells (CD3+CD56+), neutrophils (CD66b+CD45+), and macrophages (CD14+CD45+) were not significantly different in late aGVHD or cGVHD (Figure 2A-B; supplemental Table 6).
Numerous plasma markers were evaluated that have previously been associated with cGVHD or late aGVHD7,9,10,21 (Figure 2A; supplemental Table 3). Significant increases were seen at day 100 in the cGVHD cohort for ST2 and sCD13. No significant association with cGVHD could be found for CXCL9, CXCL10, CXCL11, MMP3, ICAM-1, osteopontin, interleukin IL-2Rα (IL-2Rα), soluble B-cell activating factor, Reg3α, or Tim3. No cytokine was associated with late aGVHD.
Regulatory populations can reside in the T-cell, B-cell, NK-cell, platelet, and macrophage compartments.38 B cells, associated with having regulatory function, are usually identified functionally by IL-10 production in a CD19+CD38hiCD24hi27 population and our studies were not designed to evaluate functional characteristics. We phenotypically evaluated both Treg-cell and CD56bright NKreg-cell populations (Figure 2; supplemental Table 4). We found no association between Treg-cell populations and cGVHD. There was, however, a very strong association between noncytolytic CD56bright NKreg cells (expressing CD335 and lacking cytoplasmic perforin or granzyme B) and cGVHD. None of these NKreg populations correlated with development of late aGVHD. An alternate comparison of patients in the cGVHD group vs the late aGVHD group (Figure 2C) confirmed that cGVHD compared with late aGVHD patients had decreases in NKreg cells and B cells, with increases in naive Tc cells, Th cells, and sCD13.
Impact of previous aGVHD on day 100 markers that identify those at risk of late aGVHD and cGVHD
In the previous analyses, patients without cGVHD were used as a control group regardless of previous aGVHD. In these analyses, we separated the control group without cGVHD into 2 separate controls groups: (1) those with aGVHD, or (2) those without prior aGVHD. The group that had neither cGVHD nor previous aGVHD were defined as an immune-tolerant cohort (Figure 1; n = 87). Comparison of late aGVHD to the immune-tolerant cohort identified a number of additional B-cell associations for late aGVHD, including an increase in unswitched memory B cells and T3 transitional B cells (Figure 3A; supplemental Table 5). A further analysis of the impact of previous aGVHD on the late aGVHD immune profile compared the late aGVHD cohort to the non-cGVHD control population that had had previous aGVHD (resolved by day 100; Figure 1; n = 45) and found no difference in the pattern (Figure 3B).
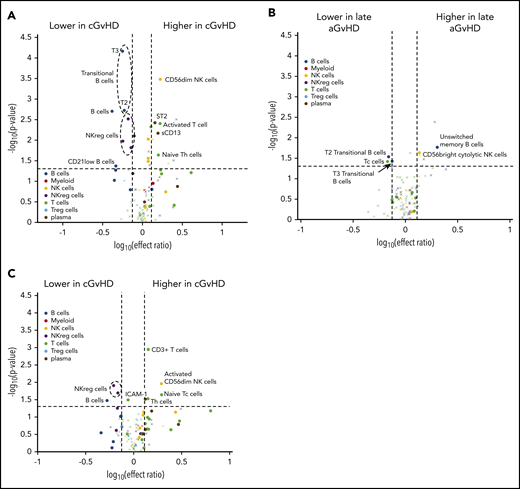
Evaluation of the impact of previously resolved early aGVHD on immune profiles at day 100 in patients who will develop late aGVHD or cGVHD. Volcano plots showing significant markers that meet all 3 criteria: P ≤ .05 (y-axis), ROC AUC ≥0.60 (circle, ≥0.60; cross, <0.6), and effect ratio ≥1.3 or ≤0.75 (x-axis). The subfigures correspond to (A) active late aGVHD compared with tolerant patients, (B) late aGVHD compared with patients who has clearly resolved aGVHD, (C) cGVHD compared with tolerant patients, and (D) cGVHD compared with patients with previous resolved early aGVHD. Cell population are identified by color, with dark blue representing B cells, orange myeloid populations, yellow NK cells, purple NKreg cells, green T cells, light blue Treg cells, and dark red plasma cytokines. We note the following clinical variables were modeled as confounding factors in the logistic regression model: (1) prophylaxis or treatment with either alemtuzumab or ATG, (2) prophylaxis or treatment with rituximab, (3) recipient age, (4) the use of a peripheral blood donor product or not, and (5) whether the donor was HLA-identical or not.
Evaluation of the impact of previously resolved early aGVHD on immune profiles at day 100 in patients who will develop late aGVHD or cGVHD. Volcano plots showing significant markers that meet all 3 criteria: P ≤ .05 (y-axis), ROC AUC ≥0.60 (circle, ≥0.60; cross, <0.6), and effect ratio ≥1.3 or ≤0.75 (x-axis). The subfigures correspond to (A) active late aGVHD compared with tolerant patients, (B) late aGVHD compared with patients who has clearly resolved aGVHD, (C) cGVHD compared with tolerant patients, and (D) cGVHD compared with patients with previous resolved early aGVHD. Cell population are identified by color, with dark blue representing B cells, orange myeloid populations, yellow NK cells, purple NKreg cells, green T cells, light blue Treg cells, and dark red plasma cytokines. We note the following clinical variables were modeled as confounding factors in the logistic regression model: (1) prophylaxis or treatment with either alemtuzumab or ATG, (2) prophylaxis or treatment with rituximab, (3) recipient age, (4) the use of a peripheral blood donor product or not, and (5) whether the donor was HLA-identical or not.
A similar comparison for cGVHD was performed to evaluate impact of previously resolved aGVHD on the immune profiles. Comparison of the cGVHD cohort to the immune-tolerant cohort (Figure 3C) demonstrated a pattern similar to that seen when compared with the entire control no-cGVHD population (Figure 2A). cGVHD markers maintained significance, regardless of prior aGVHD, including increased activated classic (CD56dim) NK cells and ST2 and decreased transitional T3 B cells, CD21low B cells, and NKreg cells (Figure 3D). One marker that was not previously significant, Reg3α, met the criteria (Figure 3D).
Differences between late aGVHD and active cGVHD between days 100 and 114 and patients at risk for future late aGVHD and cGVHD
A small cohort of children who had either active late aGVHD or active cGVHD diagnosed before day 114 were excluded from the previous analyses. We evaluated for different patterns in patients who had active late aGVHD between day 100 and day 114 compared with either the entire control population (Figure 4A, no cGVHD with or without previous aGVHD) or the immune-tolerant cohort (Figure 4B, no aGVHD or later cGVHD). In both comparisons, patients had an increase in sCD13 and ST2) and a decrease in overall B-cell numbers, transitional B cells, immunoglobulin D (IgD)+CD27+ B cells, and CD31+ naive Treg cells (Figure 4A-B). This was significantly different compared with patients who would later develop late aGVHD (Figure 2B).
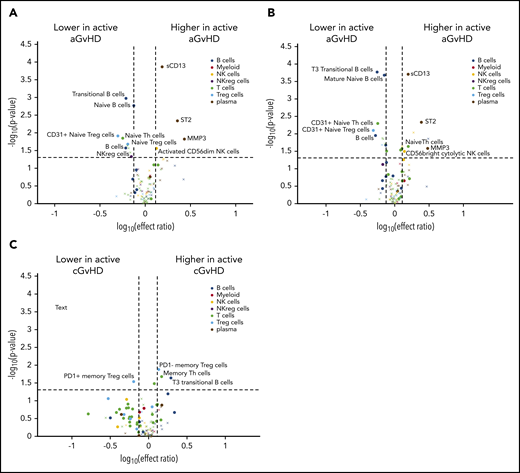
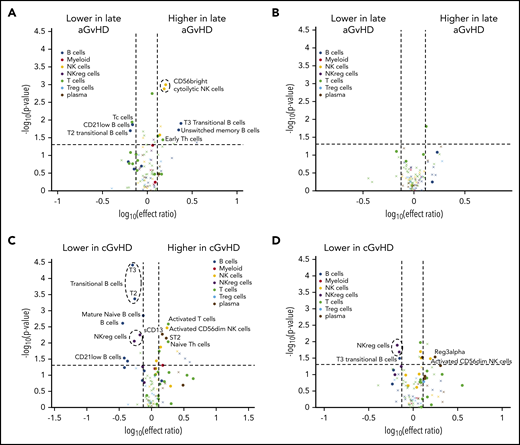
Differences between active late aGVHD and active cGVHD between days 100 and 114 and patients at risk for future late aGVHD. Volcano plots showing significant markers that meet all 3 criteria of a P ≤ .05 (y-axis), ROC AUC of ≥0.60 (circle, ≥0.60; cross, <0.6), and effect ratio ≥1.3 or ≤0.75 (x-axis). The subfigures correspond to (A) active late aGVHD compared with patients that have no later cGVHD, (B) active late aGVHD compared with tolerant patients with no cGVHD and no previous aGVHD, (C) active cGVHD that occurred before day 114 compared with the cGVHD cohort (same as in Figure 2A) that will develop cGVHD after day 114. Cell population are identified by color, with dark blue representing B cells, orange myeloid populations, yellow NK cells, purple NKreg cells, green T cells, light blue Treg cells, and dark red plasma cytokines. We note the following clinical variables were modeled as confounding factors in the logistic regression model: (1) prophylaxis or treatment with either alemtuzumab or ATG, (2) prophylaxis or treatment with rituximab, (3) recipient age, (4) the use of a peripheral blood donor product or not, and (5) whether the donor was HLA-identical or not.
Differences between active late aGVHD and active cGVHD between days 100 and 114 and patients at risk for future late aGVHD. Volcano plots showing significant markers that meet all 3 criteria of a P ≤ .05 (y-axis), ROC AUC of ≥0.60 (circle, ≥0.60; cross, <0.6), and effect ratio ≥1.3 or ≤0.75 (x-axis). The subfigures correspond to (A) active late aGVHD compared with patients that have no later cGVHD, (B) active late aGVHD compared with tolerant patients with no cGVHD and no previous aGVHD, (C) active cGVHD that occurred before day 114 compared with the cGVHD cohort (same as in Figure 2A) that will develop cGVHD after day 114. Cell population are identified by color, with dark blue representing B cells, orange myeloid populations, yellow NK cells, purple NKreg cells, green T cells, light blue Treg cells, and dark red plasma cytokines. We note the following clinical variables were modeled as confounding factors in the logistic regression model: (1) prophylaxis or treatment with either alemtuzumab or ATG, (2) prophylaxis or treatment with rituximab, (3) recipient age, (4) the use of a peripheral blood donor product or not, and (5) whether the donor was HLA-identical or not.
Patients with active cGVHD before day 114 had been excluded from all previous analysis presented above (Figure 2A,C). Since evaluation of the cell populations at day 100 correlated with later development of cGVHD, we evaluated whether those who already had the diagnosis of cGVHD at day 100 (between days 60 and 114 after transplant) demonstrated additional marker abnormalities (Figure 4C). We found that the small cohort of patients (n = 10) who already had active cGVHD at day 100 compared with those who developed cGVHD after day 114 post-HSCT showed an increase in transitional T3 B cells, memory Th cells, and PD-1− memory Treg cells. There was a concomitant decrease in PD-1+ memory Treg cells.
Evaluation of the impact of age and time of onset on the day 100 immune profiles in patients at risk for future cGVHD
We evaluated the impact of either age or the time of onset of cGVHD on the day 100 biomarker patterns. We divided each of the subject groups (cGVHD and controls) at their respective median age and restricted our analysis to the markers that were significant in the initial analysis (Figure 2A). We found that the decrease in transitional B-cell populations and elevation in ST2 and sCD13 were identical in the younger (solid circles) and older patients (open circles; Figure 5A). By contrast, the older population no longer had an increase in CD56dim NK cells, activated T cells, or naive T cells or a decrease in NKreg cells and CD21low B cells (Figure 5A). Evaluation of the impact of age on late aGVHD found that the T2 transitional B-cell population was significantly decreased in the older population only (AUC = 0.67; P = .04; effect ratio = 0.59), and the nonswitched memory B-cell population was significantly increased only in the younger group (ROC AUC = 0.75; P = .002; effect ratio = 2.3). T3 transitional B cells, Tc cells, and CD56bright cytolytic NK cells were not significant in the subanalysis by age.

Impact of age and time on immune profiles at day 100 of patients that went on to develop cGVHD. (A) Evaluation of the impact of the recipient age on the day-100 immune profile. Volcano plots showing significant markers that meet all 3 criteria of a P ≤ .05 (y-axis), ROC AUC ≥0.60 (circle, ≥ 0.60; cross, <0.6), and effect ratio ≥1.3 or ≤0.75 (x-axis). Cell populations are identified by color, with dark blue representing B cells, orange myeloid populations, yellow NK cells, purple NKreg cells, green T cells, light blue Treg cells, and dark red plasma cytokines. We divided each of the subject groups (cGVHD and controls) at their respective median age (∼12.5 years and 9 years) and repeated the above analysis for younger cGVHD vs younger controls. We restricted our analysis to the markers that were significant in the initial analysis (Figure 1A). The younger age group is represented by a solid circle the older age group by an open circle. (B) Evaluation of the impact of the time of onset of cGVHD on the day-100 immune profile. To examine the effect of cGVHD onset time, we divided the cGVHD subjects into 2 onset groups. The early-onset group (solid circles) was less than or equal to the median onset time, and the later onset group (open circles) was greater than the median onset time (166 days after HSCT). We contrasted each onset group against the group that did not develop cGVHD. (C) Group difference visualization with t-SNE. Each patient is represented as a point, where patients with similar marker values are projected onto similar locations in the t-SNE plot. Patients who later developed cGVHD are in blue, those with late aGVHD are in yellow, and non-cGVHD controls are in orange. The ellipses correspond to 1 standard deviation of from the mean of each group.
Impact of age and time on immune profiles at day 100 of patients that went on to develop cGVHD. (A) Evaluation of the impact of the recipient age on the day-100 immune profile. Volcano plots showing significant markers that meet all 3 criteria of a P ≤ .05 (y-axis), ROC AUC ≥0.60 (circle, ≥ 0.60; cross, <0.6), and effect ratio ≥1.3 or ≤0.75 (x-axis). Cell populations are identified by color, with dark blue representing B cells, orange myeloid populations, yellow NK cells, purple NKreg cells, green T cells, light blue Treg cells, and dark red plasma cytokines. We divided each of the subject groups (cGVHD and controls) at their respective median age (∼12.5 years and 9 years) and repeated the above analysis for younger cGVHD vs younger controls. We restricted our analysis to the markers that were significant in the initial analysis (Figure 1A). The younger age group is represented by a solid circle the older age group by an open circle. (B) Evaluation of the impact of the time of onset of cGVHD on the day-100 immune profile. To examine the effect of cGVHD onset time, we divided the cGVHD subjects into 2 onset groups. The early-onset group (solid circles) was less than or equal to the median onset time, and the later onset group (open circles) was greater than the median onset time (166 days after HSCT). We contrasted each onset group against the group that did not develop cGVHD. (C) Group difference visualization with t-SNE. Each patient is represented as a point, where patients with similar marker values are projected onto similar locations in the t-SNE plot. Patients who later developed cGVHD are in blue, those with late aGVHD are in yellow, and non-cGVHD controls are in orange. The ellipses correspond to 1 standard deviation of from the mean of each group.
To examine the effect of cGVHD onset time, we divided the cGVHD subjects into early-onset (≤166 days after HSCT; solid circles) and later-onset (open circles) groups. There were no major differences between the 2 groups (Figure 5B), except that ST2 and sCD13 did not meet criteria for early cGVHD, and NKreg cells had a poor correlation with later cGVHD.
t-SNE on day 100 immune profiles in patients at risk for future late aGVHD and cGVHD
To visualize differences between subject groups, we applied t-SNE to marker values of the relevant markers across all subjects. t-SNE is an unsupervised dimension reduction method that enables projection of the high-dimensional marker data onto a lower-dimensional subspace, where subjects with similar marker values would be clustered together. As shown in Figure 5C, cGVHD and controls tend to cluster in opposite side of the t-SNE plot, with late aGVHD bridging between these 2 groups.
Discussion
The ABLE/PBMTC 1202 study is the first large comprehensive study to provide data regarding the similarities and differences underlying the pathobiology of cGVHD and late aGVHD in humans. Strengths of this study include the highly standardized and reproducible central laboratory analysis of cellular and plasma markers associated with GVHD ≥3 months after HSCT, along with a thorough adjudication of GVHD features using the NIH-CC.24 The comprehensive analyses of 71 T-cell, B-cell, NK-cell, dendritic-cell, and myeloid immune populations and 12 cGVHD plasma markers allowed us to establish immune patterns for both late aGVHD and cGVHD measured either before (Figures 2 and 3) or after (Figure 4) the onset of disease. Our unsupervised dimension reduction analysis in the t-SNE plot suggests a progression from no GVHD to late aGVHD transitioning to cGVHD (Figure 5C). These analyses demonstrate that late aGVHD and cGVHD share common immune profile of abnormalities in transitional B-cell loss and increased cytolytic NK cells (Figure 6). cGVHD is more complex, with additional cytokine, T-cell, NK-cell, and B-cell abnormalities. The association of early aGVHD with the development of later cGVHD is well established.38 We observed that early resolved aGVHD had a major influence on the immune profile of late aGVHD but little influence on the abnormalities seen in cGVHD.
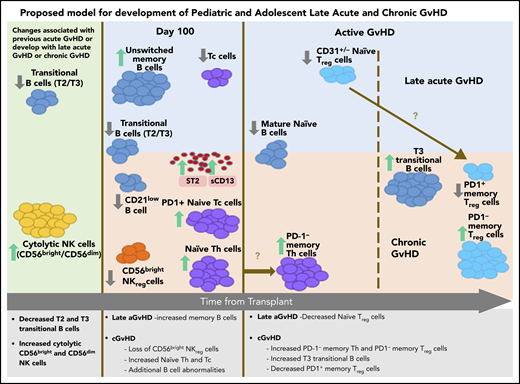
Summary of late aGVHD and cGVHD immune profile differences and progression in pediatric and adolescent patients.
Summary of late aGVHD and cGVHD immune profile differences and progression in pediatric and adolescent patients.
The loss of regulator-cell function appeared to be one of the critical events in development of cGVHD compared with late aGVHD (Figure 6). A major factor that appeared to associate with future development of cGVHD was a loss of NKreg cells and an increase in naive Th cells at day 100 (Figures 2 and 6). A loss of Treg-cell populations was seen in both active late aGVHD (CD31+/− naive Treg cells) and active cGVHD (loss of PD-1+ memory Treg cells and increase in PD-1− memory Treg cells). We suggest (Figure 6) that there is a progression of naive Th cells before the onset of cGVHD into PD-1− naive Th cells at cGVHD onset and that naive Treg cells seen in late aGVHD may mature into memory Treg cells with progression to cGVHD.
The additional loss of PD-1+ memory Treg cells and a concomitant increase in PD-1− memory Treg cells (Figure 4C) is consistent with a murine study in PD-1 knockout mice,39 where a lack of PD-1 expression resulted in a decreased extrathymic differentiation of peripheral Treg cells, supporting PD-1 dependence in peripheral Treg-cell development. The association of NKreg cells with the lack of future cGVHD development (Figure 2A) and the loss of PD-1+ memory Treg cells with existing cGVHD (Figure 4C) suggest a pattern similar to placental tolerance, where the NKreg-cell population predominates in the first trimester, followed by later expansion of Treg cells40 in the second trimester.
While aGVHD has known cytokine abnormalities, including ST2 and Reg3α,41 we identified no cytokine abnormalities in patients who would develop late aGVHD after day 114 (Figure 3A-B), although ST2 and sCD13 were elevated in active late aGVHD (Figure 4A-B), the same as in cGVHD (Figure 2A). By contrast, sCD13 was the only cytokine specific for later development of cGVHD after day 114 (Figure 2C).
The loss of NKreg-cell numbers in the development of cGVHD has been previous described.7,8 As part of the loss of CD56bright noncytolytic NKreg cells in cGVHD, we observed a concomitant increase in cytolytic CD56bright and CD56dim NK cells (Figure 2B). This is consistent with a possible model of NK progression, where CD56bright noncytolytic NK cells change toward cytolytic CD56bright and CD56dim NK effector cells through STAT3 regulation.42
We found that NIH-CC cohort and non-NIH-CC cohort patients had identical immune profiling patterns, suggesting that the NIH-CC criteria may be too specific and may require modifications in the next version of the NIH consensus diagnostic criteria. Previous resolved aGVHD impacted immune profiles at day 100 in patients at risk for both late aGVHD and cGVHD. This was particularly true for late aGVHD, where the differences were primarily identified when compared with only the tolerant control group.
One age-related difference we observed was that CD21low B cells were lower in children with cGVHD compared with the increases observed in adults with cGVHD.22 This was surprising, as our group had confirmed the increase of CD21low B cells in cGVHD in adults as previously reported.11,12,22 Our comparison of the CD21low B cells in younger vs older patients (Figure 5A) found a loss of a significant decrease in the older populations, suggesting that age may impact how this population functions. It is possible this was seen due to the relatively high number of donor marrow transplants in children compared with a preponderance of mobilized peripheral blood in adult HSCT, although the results were adjusted for clinical variables, including peripheral blood donor product. There are no previously identified age-related differences in CD21low B-cell populations,43 and evaluation of the underlying biology is needed.
The comprehensive immune and cytokine profile approach used in this multicenter study will allow us to better risk-assign HSCT patients into low- and high-risk groups for the later development of cGVHD and late aGVHD while developing targeted therapeutic approaches based on the biological patterns identified. Biomarker algorithms will assist in assigning patient risk for cGVHD, with the possibility of rapid withdrawal of immunosuppressive prophylaxis in low-risk populations and early interventions using novel pre-emption for high-risk populations, aimed at minimizing the future development of cGVHD. A limitation of this study is the restriction to children and adolescents. ABLE validation studies are occurring in patients ≤30 years, although similar studies are needed in older adults.
The online version of this article contains a data supplement.
The publication costs of this article were defrayed in part by page charge payment. Therefore, and solely to indicate this fact, this article is hereby marked “advertisement” in accordance with 18 USC section 1734.
Acknowledgments
The authors thank the PBMTC network for support.
A Canadian Institutes of Health Research team grant funded these studies. The team grant received funding from the Canadian Institutes of Health Research, the Canadian Cancer Society, the C17 Research Network, the Garron Family Cancer Center at the Hospital for Sick Children, and the Pediatric Oncology Group of Ontario. Support for PBMTC efforts was provided by a grant from the Johnny Crisstopher Children’s Charitable Foundation St. Baldrick’s Consortium as well as National Institutes of Health, National Heart, Lung, and Blood Institute/National Cancer Institute grant 2UG1HL069254.
Authorship
Contribution: K.R.S. designed the studies, wrote the manuscript, and oversaw of all aspects of the trial and evaluation of the results; A.K. performed and interpreted all of laboratory studies, wrote the manuscript, and evaluated the results; B.N. performed all statistical analyses; S.A. and M.L. performed and interpreted the laboratory studies; E.R.N., J.T.W., C.L.K., V.A.L., T.S., D.A.J., and A.C.H. contributed study subjects, adjudicated on clinical classification, wrote the manuscript, and interpreted results; M.A.P. contributed the PBMTC infrastructure and study subjects, adjudicated on clinical classification, wrote the manuscript, and interpreted results; H.B., S.W.C., E.H.C., K.A.K., M.B., B.R.O., A.F., S.C., D.C., J.H.C., M.J., S.S., A.B.P., G.C.M., D.M., A.C.C., and A.L. contributed study subjects, wrote the manuscript, and interpreted results; S.A. performed assays; E.O. interpreted data and wrote the manuscript; P.S. managed aspects of the study coordination and wrote the manuscript; A.H. managed all aspects of study management and coordination and wrote the manuscript; S.M. supervised all statistical analyses and data interpretation; and G.D.E.C. designed the studies, wrote the manuscript, oversaw all clinical aspect of the trial, and evaluated the results.
Conflict-of-interest disclosure: The authors declare no competing financial interests.
Correspondence: Kirk R. Schultz, BC Children’s Hospital and Research Institute, 950 West 28th Ave, Room 3102, Vancouver, BC V5Z 4H4, Canada; e-mail: kschultz@mail.ubc.ca.

This feature is available to Subscribers Only
Sign In or Create an Account Close Modal