

In this issue of Blood, characterize clonal hematopoiesis in serial samples from persons with mantle cell lymphoma (MCL) undergoing frontline treatment, providing evidence that treatment plays an important role in the development of clonal hematopoiesis.1
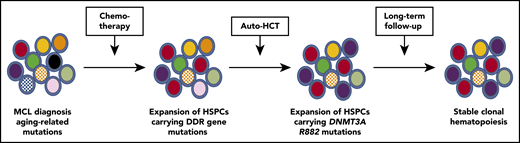
Evolution of clonal hematopoiesis during treatment of persons with MCL. HSPCs acquire somatic mutations with aging, resulting in the production of a genetically heterogenous pool of HPSCs. Genotoxic stress induced by chemotherapy favors the selection of HSP-Cs carrying mutations of DNA damage response genes, such as TP53 and PPM1D (red cells). Auto-HCT induces replicative stress and potentially other stresses related to an altered bone marrow microenvironment. In this study, auto-HCT was associated with the selective expansion of HPSCs carrying DNTM3A R882 mutations (purple cells). During long-term follow-up, the existing clonal hematopoiesis was mostly stable.
Evolution of clonal hematopoiesis during treatment of persons with MCL. HSPCs acquire somatic mutations with aging, resulting in the production of a genetically heterogenous pool of HPSCs. Genotoxic stress induced by chemotherapy favors the selection of HSP-Cs carrying mutations of DNA damage response genes, such as TP53 and PPM1D (red cells). Auto-HCT induces replicative stress and potentially other stresses related to an altered bone marrow microenvironment. In this study, auto-HCT was associated with the selective expansion of HPSCs carrying DNTM3A R882 mutations (purple cells). During long-term follow-up, the existing clonal hematopoiesis was mostly stable.
Clonal hematopoiesis is characterized by an expanded population of hematopoietic stem/progenitor cell (HSPC) clones carrying 1 or more somatic mutations. Its incidence increases with age and prior exposure to myeloablative therapy. However, the size of the mutant HSPC clone is often stable over time, suggesting that most mutations confer, at best, a modest cell intrinsic advantage to HSPCs. This is consistent with mouse studies showing that heterozygous mutations of common clonal hematopoiesis genes, including DNTM3A, TET2, ASXL1, and TP53, do not provide a competitive advantage after transplantation. How then do HSPCs carrying these mutations expand sufficiently to produce detectable clonal hematopoiesis? Recent data suggest that intermittent environmental stressors may play a role. For example, we and others recently showed that exposure to myeloablative therapy results in the selective expansion of HSPCs carrying mutations in DNA repair genes, including TP53 and PPM1D.2-4 There also is evidence that inflammatory stress results in the expansion of Tet2-deficient HSPCs in mice.5
Eskelund et al, in this issue of Blood, characterized clonal hematopoiesis in younger persons (age <66 years) with MCL receiving frontline treatment that included consolidation with autologous hematopoietic cell transplantation (auto-HCT) after induction chemotherapy. Bone marrow or blood samples were available for 149 persons achieving minimal residual disease–negative remission following auto-HCT. Samples also were available in a subset of patients at the time of MCL diagnosis (prior to any chemotherapy), after induction immunochemotherapy, and after long-term follow-up after completing treatment (median 54 months). All samples were analyzed using an error-corrected sequencing approach to detect mutations in a panel of 22 genes implicated in clonal hematopoiesis with a variant allele frequency cutoff of 1%. Clonal hematopoiesis was detected in 30% of cases after auto-HCT, with a total of 54 mutations identified. Although the sample size was small, longitudinal analysis of these mutations support the following conclusions (see figure). First, the majority of mutations present after auto-HCT were detectable prior to any treatment, suggesting expansion of preexisting HSPCs carrying aging-related mutations rather than de novo mutation generation during treatment. Second, consistent with prior reports, HSPCs carrying mutations in DNA repair genes selectively expanded following induction chemotherapy but did not did not further expand after auto-HCT or during long-term follow-up. Finally, a modest but significant expansion of HSPCs carrying clonal hematopoiesis mutations was observed following auto-HCT. This was mostly due to HSPCs carrying likely driver mutations other than DNA damage response gene mutations.
The research adds to accumulating evidence that environmental stressors contribute to the development of clonal hematopoiesis. Genotoxic stress induces a p53-dependent growth arrest, which is attenuated in HSPCs carrying DNA damage response gene mutations, conferring a selective advantage to these HSP-C clones. HCT induces several potential stressors that might affect HSCs, including the need for HSCs to rapidly proliferate following transplantation and disruption of the stem niche that occurs after myeloablative conditioning for HCT. Prior studies reported the rapid expansion of clonal hematopoiesis after HCT in ∼50% of cases.2,6 Consistent with the current study, HSPCs carrying DNA damage response genes do not consistently expand following HCT. On the other hand, although the evidence is still largely anecdotal, DNTM3A R882 mutations appear to confer a strong selective advantage following HCT.2
There remain many open questions in the field. Are there other types of environmental stressors that contribute to the development, maintenance, or progression of clonal hematopoiesis? Inflammation appears to contribute to the clonal expansion of HSPCs carrying mutations in epigenetic modifies, such as TET2 and DNMT3A. However, the impact of the type, severity, or duration of inflammation on clonal hematopoiesis needs to be defined. For example, the impact of changes in the gut microbiome on clonal hematopoiesis is uncertain. There is an increase in the use of immunotherapies for malignancies and inflammatory conditions. Do these therapies and therapies that target metabolism (for example) select for HSPC clones carrying specific mutations? Finally, it will be important to determine whether environmental stressors play a role in the evolution of clonal hematopoiesis to hematopoietic malignancy. To address these and related questions, it will be important to broadly survey the genome for mutations, rather than focus on mutations commonly mutated in aging-related clonal hematopoiesis. This point is illustrated by the frequent development of clonal hematopoiesis due to mutations in PIGA and BCOR/BCORL1 in patients with aplastic anemia.7 Most clonal hematopoiesis studies focus on single nucleotide variants. However, clonal hematopoiesis due to chromosomal abnormalities, such as copy number alterations, also is common.8 The impact of environmental stressors on the selection of HSPCs carrying structural variants is largely unknown. Platforms with a high-throughput capacity for integrating copy number variation, translocations, and single nucleotide variation will allow us to form a more complete picture of clonal hematopoiesis in the context of therapy-related and other environmental stressors.
Conflict-of-interest disclosure: The author declares no competing financial interests.

This feature is available to Subscribers Only
Sign In or Create an Account Close Modal