

Key Points
PSplt controls platelet activation as well as coagulation in thrombi in large veins, but not in large arteries.
PSplt limits thrombin generation in the thrombus and ensures that highly activated platelets and fibrin remain localized at the injury site.

Abstract
Anticoagulant protein S (PS) in platelets (PSplt) resembles plasma PS and is released on platelet activation, but its role in thrombosis has not been elucidated. Here we report that inactivation of PSplt expression using the Platelet factor 4 (Pf4)-Cre transgene (Pros1lox/loxPf4-Cre+) in mice promotes thrombus propensity in the vena cava, where shear rates are low, but not in the carotid artery, where shear rates are high. At a low shear rate, PSplt functions as a cofactor for both activated protein C and tissue factor pathway inhibitor, thereby limiting factor X activation and thrombin generation within the growing thrombus and ensuring that highly activated platelets and fibrin remain localized at the injury site. In the presence of high thrombin concentrations, clots from Pros1lox/loxPf4-Cre− mice contract, but not clots from Pros1lox/loxPf4-Cre+ mice, because of highly dense fibrin networks. Thus, PSplt controls platelet activation as well as coagulation in thrombi in large veins, but not in large arteries.
Introduction
Protein S (PS), named after Seattle, Washington, the city of its discovery, was first reported as a vitamin K-dependent plasma glycoprotein in 1977.1 PS functions as a cofactor for activated protein C (APC) in the regulation of activated factor V (FV) and FVIII2-6 and for tissue factor pathway inhibitor (TFPI),7 as well as in the inhibition of activated FIX.8
The crucial anticoagulant role of PS is demonstrated by the severe clinical manifestations presented by patients with homozygous and compound heterozygous severe PS deficiency.9-11 Purpura fulminans and disseminated intravascular coagulation (DIC) in the neonatal period are observed in these patients.
Heterozygous PS deficiency, first reported in 1984,10,11 has milder clinical consequences, but is associated with an increased risk for venous thromboembolism (VTE).12
Most of the insights regarding the association between PS deficiency and VTE come from studies of families carrying inherited thrombophilia. This brought the notion that the risk for VTE in PS-deficient subjects is 5 to 10 times higher than in non-PS-deficient relatives.12-15 The clinical relevance for the individual patient is important, as subjects with familial PS deficiency have a first VTE incidence of 0.7% per year16 and an annual recurrence risk of 6% to 10%.17-19 Moreover, in population-based studies such as the Multiple Environmental and Genetic Assessment (MEGA) of risk factors for venous thrombosis) case-control study20 and the Leiden Thrombophilia Study,21 low levels of free PS were associated with an increased risk for thrombosis.
PS that circulates in blood derives principally from hepatocytes and vascular endothelial cells. In addition, about 2.5% of circulating PS is found within platelet α-granules.22,23 Differently from human FV that is derived primarily or exclusively via uptake from the plasma pool,24 Platelet PS (PSplt) is derived exclusively from expression within the megakaryocytes. Although platelet granule release happens during thrombus formation and platelets are the main cellular constituent of a clot, little is known about the functionality and importance of PSplt. To uncover the in vivo role of PSplt, we generated and studied a mouse model lacking PS expression in platelets.
Materials and methods
Mice
C57BL/6-inbred Platelet factor 4 (Pf4)-Cre+ mice were obtained and genotyped as described [C57BL/6-Tg(Pf4-icre)Q3Rsko/J; The Jackson Laboratory]. 129Sv-C57BL/6J Pros1 transgenic mice containing 2 loxP sites between exons 3 and 7 (Pros1lox/lox) were generated through conditional gene targeting, using the Cre-loxP system as described.25 Pros1lox/lox mice were bred with Pf4-Cre+ mice to inactivate the Pros1lox/lox gene in the megakaryocyte lineage. Pros1lox/lox mice were crossed with Pros1+/− to obtain Pros1lox−-, as heterozygous Pros1 control for the transgenic mice used in this study.25 Multiplex polymerase chain reaction (PCR) to allow the amplification of the inserted lox sequence in both Pros1 alleles and of the Pros1 null allele was performed as described.25 Male and female littermates aged 8 to 16 weeks were used throughout the study. The Swiss Federal Veterinary Office approved the experiments.
FeCl3-injury thrombosis model in mesenteric arterioles
A model of thrombosis in mesenteric arterioles using intravital microscopy was applied according to Prince et al.26
For some histological investigations, a mixture of thrombin/Xa-specific fluorogenic substrate (Z-Gly-Gly-Arg-AMC, 4002155; Bachem; 1/125 vol/vol) and fluorescein isothiocyanate conjugated anti-CD41 (MWReg30; BD Pharmingen; 2 µg/body weight) antibody was injected via retroorbital sinus 5 minutes before FeCl3 injury.
For immunostaining, FeCl3-injured mesenteric arterioles were fixed in paraformaldehyde (PFA) 4%. The following antibodies were used: anti-fibrin (final concentration, 15.6 µg/mL; monoclonal antibody clone, 102-10)27 with Alexa 568-conjugated goat anti-human as secondary antibody, diluted 1:500; BV711-conjugated anti-CD62 (final concentration, 10 µg/mL; clone RB40.34; BD Pharmingen); and fluorescein isothiocyanate-conjugated anti-CD41 (final concentration, 10 µg/mL; MWReg30; BD Pharmingen).
Staining was examined using the Zeiss LSM710 Laser scanning microscope for z stacks and EC Plan-Neofluar 20×/1.30 Oil DIC M27/a = 0.21 mm as objective. Images were acquired and optimized with ZEN system software. Imaris software (Bitplane) was used for visualization of 3-dimensional confocal data, volume, and surface rendering.
Vena cava and carotid artery thrombus formation monitoring using ultrahigh-frequency ultrasound imaging system
The thrombosis models in the inferior vena cava and in the carotid artery were performed according to Aghourian et al28 and Novotny et al,29 with minor modifications. To induce thrombus formation, a Whatman filter paper (4 × 2 mm) saturated in 10% FeCl3 was applied to the vessels and removed after 3 minutes. All mice underwent ultrahigh-frequency ultrasound (US) imaging at different points: before FeCl3 application to the vessel and 5, 10, 20, and 30 minutes after injury.
A Vevo 3100 ultrahigh-frequency US imaging system (Fujifilm VisualSonics) and a MX550S linear array transducer (bandwidth, 32-55 MHz; center frequency, 40 MHz) were used to acquire US videos and volumes.
Thrombus formation, blood flow dynamics, and detailed flow profile were reconstructed in 2 and 3 dimensions by color Doppler and pulse-wave Doppler, electrocardiogram-triggered high-frequency US imaging modes in inferior vena cava. Three- and 4-dimensional US image series were acquired at 76-µm intervals. The images and videos were analyzed using a VevoLab software package (Fujifilm VisualSonics).
After US imaging, the vessels were ligated, fixed in PFA 4%, and embedded in paraffin. Serial sections (2.5 µm) with no pretreatment were stained with hematoxylin/eosin or immunostained for insoluble fibrin or PS. The following antibodies were used: anti-fibrin (monoclonal antibody clone 102-1027 ; final concentration, 5.95 µg/mL) and secondary antibody rabbit anti-human (ab7155; Abcam; 1:300), PS (MAB 4976; R&D; 1:50) and secondary antibody rabbit anti-rat (1:200; ab102248; Abcam), and CD31 (ab28364; Abcam; 1:30) and secondary antibody (Bond Polymer Refine Detection, Leica Biosystems; DS9800). Whole slides were scanned using 3-dimensional HISTECH Panoramic 250 Flash II, with 20× (NA 0.8) and 40× (NA 0.95) air objectives.
Some ligated vessels were prepared for section electron microscopy. After fixation in 2.5% glutaraldehyde solution, vessels were washed with 0.15 M HEPES 2 times for 10 minutes and dehydrated in 70%, 80%, and 96% ethanol (Alcosuisse) for 30 minutes each at room temperature. Subsequently, vessels were immersed in 100% ethanol (Merck) 3 times for 30 minutes. They were critical point dried with a CPD 300 (Leica Microsystems), divided exactly into 2 pieces, and sputter coated with 10 nm of gold with a sputter coater (Balzers Union). Vessels were then examined with a scanning electron microscope (Zeiss DSM 982).
Additional methods are described in the supplemental Materials and methods, available on the Blood Web site.
Results
Generation of Pros1lox/loxPf4-Cre+ mice
To explore the role of PS in platelets, mice carrying a conditional Pros1 knockout allele in the megakaryocyte lineage were generated using the Pf4 promoter as Cre driver (Pros1lox/loxPf4-Cre+; supplemental Figure 1A). Practically, we bred Pros1lox/lox mice25 with Pf4-Cre transgenic mice30 to obtain Pros1lox/loxPf4-Cre+ mice. Pros1lox/loxPf4-Cre+ mice appeared completely normal with no difference in size, weight, or behavior compared with Pros1lox/loxPf4-Cre− mice, and were fertile. Their viability was monitored up to 16 months (n = 4) without showing any difference compared with Pros1lox/loxPf4-Cre− mice.
Pros1lox/loxPf4-Cre− and Pros1lox/loxPf4Cre+ littermates were genotyped by PCR (Figure 1A). In this model, the Cre-recombinase activity was confined to megakaryocytes.30 Therefore, as expected, Pros1lox/loxPf4-Cre+ mice did not express PS in platelets (Figure 1B and D). PS content in Pros1lox/− platelets was ∼40% to 50% less than in Pros1lox/loxPf4-Cre− platelets (Figure 1B). In contrast, PS plasma levels were comparable in Pros1lox/loxPf4-Cre− and Pros1lox/loxPf4-Cre+ mice (Figure 1C). Pros1lox/− mice displayed ∼50% to 60% less plasma PS than Pros1lox/loxPf4-Cre− and Pros1lox/loxPf4-Cre+ mice (Figure 1C), as predicted.25,31
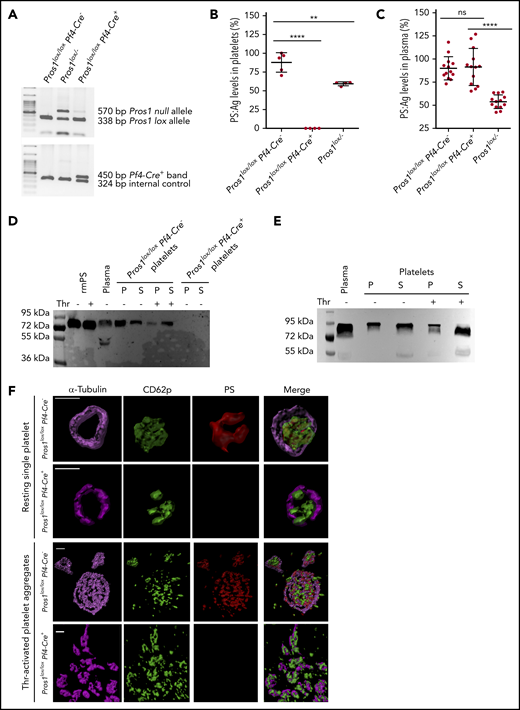
Generation of a mouse model lacking PS expression in platelets (Pros1lox/loxPf4-Cre+). (A) Genotyping results of mice with normal PS expression (Pros1lox/loxPf4-Cre−), Pros1lox/−, and Pros1lox/loxPf4-Cre+ mice using genomic DNA extracted from ear tissues. The presence of lox sequences in Pros1 alleles and of Pf4-Cre transgene was evaluated by 2 independent multiplex PCRs. PCR products were then subjected to electrophoresis. The lox band had a lower molecular weight (338 bp) compared with the null band (570 bp), in accordance with Saller et al25 ; the Pf4-Cre+ band has a higher molecular weight (450 bp) in comparison with the internal PCR control (324 bp). (B) PS antigen levels in platelet lysates from Pros1lox/loxPf4-Cre−, Pros1lox/loxPf4-Cre+, and Pros1lox/− mice. Results are expressed as mean ± standard error of the mean (SEM) of percentage relative to the pooled normal platelet lysate. **P ≤ .01; ****P ≤ .0001. (C) PS antigen levels in plasma samples from Pros1lox/loxPf4-Cre−, Pros1lox/loxPf4-Cre+, and Pros1lox/− mice. Results are expressed as mean ± SEM of percentage relative to the pooled normal plasma. ns, not significant; ****P ≤ .0001. (D-E) PS isoforms in plasma and platelet after activation by thrombin. Western blotting after sodium dodecyl sulfate-polyacrylamide gel electrophoresis under reducing conditions was performed using a monoclonal antibody raised against murine PS (D) and a polyclonal antibody against human PS (E). P, platelet; S, platelet releasate; Thr, thrombin. (F) Confocal microscopy of resting and thrombin-activated mouse Pros1lox/loxPf4-Cre− and Pros1lox/loxPf4-Cre+ platelets, stained for -tubulin (in magenta), CD62p (in green), and PS (in red). Scale bar, 2 μm. Acquired z stacks were used for volumes and surface rendering by Imaris software.
Generation of a mouse model lacking PS expression in platelets (Pros1lox/loxPf4-Cre+). (A) Genotyping results of mice with normal PS expression (Pros1lox/loxPf4-Cre−), Pros1lox/−, and Pros1lox/loxPf4-Cre+ mice using genomic DNA extracted from ear tissues. The presence of lox sequences in Pros1 alleles and of Pf4-Cre transgene was evaluated by 2 independent multiplex PCRs. PCR products were then subjected to electrophoresis. The lox band had a lower molecular weight (338 bp) compared with the null band (570 bp), in accordance with Saller et al25 ; the Pf4-Cre+ band has a higher molecular weight (450 bp) in comparison with the internal PCR control (324 bp). (B) PS antigen levels in platelet lysates from Pros1lox/loxPf4-Cre−, Pros1lox/loxPf4-Cre+, and Pros1lox/− mice. Results are expressed as mean ± standard error of the mean (SEM) of percentage relative to the pooled normal platelet lysate. **P ≤ .01; ****P ≤ .0001. (C) PS antigen levels in plasma samples from Pros1lox/loxPf4-Cre−, Pros1lox/loxPf4-Cre+, and Pros1lox/− mice. Results are expressed as mean ± SEM of percentage relative to the pooled normal plasma. ns, not significant; ****P ≤ .0001. (D-E) PS isoforms in plasma and platelet after activation by thrombin. Western blotting after sodium dodecyl sulfate-polyacrylamide gel electrophoresis under reducing conditions was performed using a monoclonal antibody raised against murine PS (D) and a polyclonal antibody against human PS (E). P, platelet; S, platelet releasate; Thr, thrombin. (F) Confocal microscopy of resting and thrombin-activated mouse Pros1lox/loxPf4-Cre− and Pros1lox/loxPf4-Cre+ platelets, stained for -tubulin (in magenta), CD62p (in green), and PS (in red). Scale bar, 2 μm. Acquired z stacks were used for volumes and surface rendering by Imaris software.
Pros1lox/loxPf4-Cre− and Pros1lox/loxPf4Cre+ bone marrow-derived monocytes/macrophages expressed a comparable PS amount, whereas PS was not detected in Pros1lox/loxPf4-Cre− and Pros1lox/loxPf4Cre+ neutrophils (supplemental Figure 1B).
Using a standard curve of murine PS, PSplt and plasma PS concentrations were measured in Pros1lox/loxPf4-Cre− mice. Similar to humans,23,32 lysates from 108 murine wild-type (WT) platelets contained 261 ± 32 ng of PS. In Pros1lox/loxPf4-Cre− mouse plasma, PS concentration was lower than in human plasma samples33 (reference range, 5.2-7.4 vs 15-25 µg/mL, respectively). However, in human plasma, 60% of PS associates noncovalently and reversibly with the complement regulatory protein C4b-binding protein, interfering with PS anticoagulant functions.34 The remaining 40%, accounting for 6.5 to 10 µg/mL of PS, circulates as free protein and is responsible for the anticoagulant APC-cofactor functions of plasma PS.33 In contrast, in mice, PS-C4b-binding protein complexes do not form because of the lack of the C4b-binding protein β-chain necessary for the binding. Therefore, only the free fully anticoagulant PS isoform circulates in murine blood.35 Taking into account the concentration of the unbound human PS, these data show that both PS content in murine platelets and PS concentration in murine plasma are comparable to those in humans.
Western blot analysis showed that murine PSplt migrates as bands with molecular weights ranging between 72 and 85 kDa, similar to the plasma isoform (Figure 1D). Once activated with thrombin, platelets released PS, displaying a band with a lower molecular weight ∼75 kDa, consistent with the cleavage of PS in the thrombin-sensitive region (Figure 1D). Comparable data were obtained when platelets were stimulated either with adenosine 5′-diphosphate (ADP) or the thromboxane A2 analog, U46619 (supplemental Figure 1C). As expected, no PS was detected in Pros1lox/loxPf4-Cre+ platelet lysates (Figure 1D).
Similar results were obtained using human samples, as reported.23 PSplt comigrates with plasma PS as bands with molecular weights ranging between 72 and 95 kDa (Figure 1E).
PSplt was localized in murine platelet α-granules, as described in human platelets.23 Indeed, PSplt colocalized with P-selectin (CD62p) not only in single Pros1lox/loxPf4-Cre− resting platelets but also in platelet aggregates generated from Pros1lox/loxPf4-Cre− thrombin-activated platelets (Figure 1F; supplemental Figure 2). In addition, as shown in supplemental Movies 1 and 2, a part of secreted PS located on the surface of platelet aggregates. As expected, PS was not detected in Pros1lox/loxPf4-Cre+ single resting platelets and platelet aggregates (Figure 1F; supplemental Figure 2).
Because activated platelets are an important source of pro- and anticoagulant factors, we also assessed the content of FV and TFPIα in Pros1lox/loxPf4-Cre− and Pros1lox/loxPf4-Cre+ platelets. Both proteins did not show expression differences in Pros1lox/loxPf4-Cre− and Pros1lox/loxPf4-Cre+ platelets (supplemental Figure 3A-B). Notably, all FV isoforms are present in murine platelets (supplemental Figure 2B). Western blotting for PS in Pros1lox/loxPf4-Cre− platelets lysates was performed before and after immunoprecipitation, using an antibody directed against TFPI or FV (supplemental Figure 3C). In addition, western blotting for TFPI in Pros1lox/loxPf4-Cre− platelets lysates was realized before and after immunoprecipitation, using an antibody directed against PS or FV (supplemental Figure 3D). These data revealed that, in mice, TFPIα and FV form a complex with PSplt.
As complete Pros1 deficiency in mice leads to consumptive coagulopathy,25,31 we assessed whether Pros1lox/loxPf4-Cre+ mice display DIC (supplemental Table 1; supplemental Figure 3). Blood analyses showed that Pros1lox/loxPf4-Cre+ mice exhibit normal blood cell counts, prothrombin and activated partial thromboplastin times, FV and fibrinogen activity levels, as well as von Willebrand factor antigen level (supplemental Table 1). In addition, DIC parameters were comparable in Pros1lox/loxPf4-Cre− and Pros1lox/loxPf4-Cre+ mice and indicated that Pros1lox/loxPf4-Cre+ did not develop overt DIC (supplemental Figure 4A). Moreover, no thrombosis or fibrin deposition was found in brain, lungs, liver, and kidney of Pros1lox/loxPf4-Cre+ mice (supplemental Figure 4B). Finally, during the whole lifespan of Pros1lox/loxPf4Cre+, neither spontaneous bleeding nor thrombosis was observed.
Thus, Pros1lox/loxPf4-Cre+ mice constitute an adequate model of PS deficiency in the megakaryocytic lineage.
Lack of PSplt does not affect platelet activation in steady state
We investigated whether Pros1lox/loxPf4-Cre+ platelets are activated in steady state. Therefore, we measured the plasma level of PF4, a protein belonging to the CXC chemokine family. This chemokine is released from α-granules of activated platelets and binds with high affinity to heparin-like molecules promoting coagulation. We found no difference in PF4 plasma level between Pros1lox/loxPf4-Cre− and Pros1lox/loxPf4-Cre+ mice. Moreover, PF4 level in both genotypes was within the normal range (Figure 2A). We confirmed the lack of platelet activation in steady state in both Pros1lox/loxPf4-Cre− and Pros1lox/loxPf4-Cre+ mice by demonstrating the absence of P-selectin (CD62p) expression at the platelet surface (Figure 2B).
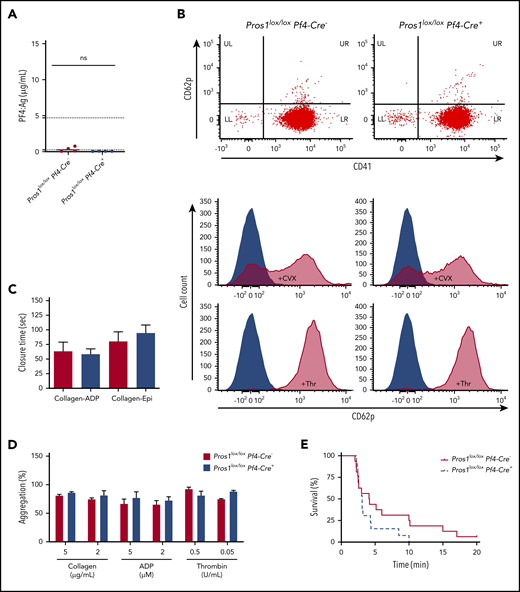
Lack of PS in platelets does not affect platelet activation state in steady state, platelet aggregation ex vivo, and platelet-dependent VTE in vivo. (A) PF4 antigen levels in plasma from Pros1lox/loxPf4-Cre− and Pros1lox/loxPf4-Cre+ mice (ns, not significant; P = .145). Data are expressed as median. The normal range of the PF4 antigen concentration in mouse plasma (0.26-4.70 µg/mL) provided by the manufacturer is indicated in the y-axis by dashed lines. (B) Flow cytometry analysis of CD62p and CD41 on resting and convulxin (10 µg/mL)-activated or thrombin (10 U/mL)-activated platelets. (C) Platelet Function Analyzer-100 closure time using collagen-ADP and collagen-epinephrine (collagen-Epi) cartridges to activate platelets in whole blood from Pros1lox/loxPf4-Cre− and Pros1lox/loxPf4-Cre+ (n = 5/genotype). Data are expressed as mean ± SEM. (D) Aggregation of washed platelets suspensions were initiated by adding different concentrations of collagen, ADP (in presence of 2 mg/mL fibrinogen), and thrombin. Results are given as mean ± SEM of at least 3 independent experiments; each of them was obtained from platelet pools (n = 4 mice/genotype). (E) Venous thromboembolism model induced by an intravenous injection of collagen-epinephrine in Pros1lox/loxPf4Cre− (straight line; n = 16) and mice Pros1lox/loxPf4Cre+ (dashed line; n = 13) mice.
Lack of PS in platelets does not affect platelet activation state in steady state, platelet aggregation ex vivo, and platelet-dependent VTE in vivo. (A) PF4 antigen levels in plasma from Pros1lox/loxPf4-Cre− and Pros1lox/loxPf4-Cre+ mice (ns, not significant; P = .145). Data are expressed as median. The normal range of the PF4 antigen concentration in mouse plasma (0.26-4.70 µg/mL) provided by the manufacturer is indicated in the y-axis by dashed lines. (B) Flow cytometry analysis of CD62p and CD41 on resting and convulxin (10 µg/mL)-activated or thrombin (10 U/mL)-activated platelets. (C) Platelet Function Analyzer-100 closure time using collagen-ADP and collagen-epinephrine (collagen-Epi) cartridges to activate platelets in whole blood from Pros1lox/loxPf4-Cre− and Pros1lox/loxPf4-Cre+ (n = 5/genotype). Data are expressed as mean ± SEM. (D) Aggregation of washed platelets suspensions were initiated by adding different concentrations of collagen, ADP (in presence of 2 mg/mL fibrinogen), and thrombin. Results are given as mean ± SEM of at least 3 independent experiments; each of them was obtained from platelet pools (n = 4 mice/genotype). (E) Venous thromboembolism model induced by an intravenous injection of collagen-epinephrine in Pros1lox/loxPf4Cre− (straight line; n = 16) and mice Pros1lox/loxPf4Cre+ (dashed line; n = 13) mice.
Lack of PSplt does not affect platelet aggregation ex vivo and platelet-dependent VTE in vivo
The specific role of PSplt in platelet function has never been investigated. Therefore, we measured the expression of P-selectin (CD62p) after platelet stimulation by convulxin or thrombin. There was no difference between Pros1lox/loxPf4-Cre− and Pros1lox/loxPf4-Cre+ mice regarding P-selectin expression (Figure 2B). We also measured the closure time assessed by the Platelet Function Analyzer-100. The closure time was similar in Pros1lox/loxPf4-Cre− and Pros1lox/loxPf4-Cre+ mice (Figure 2C). In addition, platelet aggregation studies performed on washed platelets in response to collagen, ADP, and thrombin displayed comparable results in Pros1lox/loxPf4-Cre− and Pros1lox/loxPf4Cre+ mice (Figure 2D; supplemental Figure 5).
We investigated mice survival after intravenous injection of a mixture of collagen and epinephrine, a model known to induce platelet-dependent VTE.36-38 Although there was a trend for increased mortality in Pros1lox/loxPf4Cre+ compared with Pros1lox/loxPf4Cre− mice, no significant differences were found between Pros1lox/loxPf4-Cre− and Pros1lox/loxPf4-Cre+ mice (Figure 2E).
Effect of the lack of PSplt on global hemostasis tests
To further assess whether the lack of PSplt affects hemostasis, 2 global hemostasis tests were conducted: rotation thromboelastometry (ROTEM) analysis and total thrombus-formation analysis system.
ROTEM analyzes whole blood and gives information about all stages of hemostasis process. In the extrinsic‐activated rotational thromboelastometry assay, tissue factor (TF) is used as activator. Clotting time as well as the parameters affecting the clot polymerization were similar in Pros1lox/loxPf4-Cre− mice and Pros1lox/loxPf4-Cre+ mice (Figure 3A). The extrinsically activated thromboelastometric test with cytochalasin D assay isolates fibrinogen function. By using cytochalasin D to inhibit platelets, this test blocks platelet contribution to clot formation, leaving only the coagulation proteins. The extrinsically activated thromboelastometric test with cytochalasin D parameters was similar in both genotypes (Figure 3B). Altogether, these data show that the absence of PSplt does not have an effect on the speed of whole blood clotting, and that the contribution of functional fibrinogen to clot formation is not affected by the lack of PSplt. In addition, the clot elasticity attributable to platelets (MCEplt)39 was comparable in Pros1lox/loxPf4-Cre− mice and Pros1lox/loxPf4-Cre+ mice (211 ± 10 vs 209 ± 62, respectively; P > .05).

Lack of PS in platelets affects global hemostasis assessment ex vivo and in vivo. (A-B) Whole blood activation of coagulation and clot polymerization determined by ROTEM analysis rotational (thromboelastography). (A) Extrinsic‐activated rotational thromboelastometry (EXTEM) assay, (B) extrinsically activated thromboelastometric test with cytochalasin D (FIBTEM). CT, coagulation time; CFT, clot formation time; α, α-angle; CFR, clot formation rate, MCF, maximum clot firmness; MCE, maximum clot elasticity. (C-D) Total thrombus-formation analysis on whole blood on collagen and TF-coated chip (AR-chip) at low (240 s−1, C) and high (600 s−1, D) shear rate. (E-F) Bleeding time (E) and blood loss (F) were measured after 2-mm tail transection in Pros1lox/loxPf4-Cre− (n = 7), Pros1lox/loxPf4-Cre+ (n = 7), and Pros1lox/− (n = 5) mice. All data are expressed as mean ± SEM. ns, not significant; *P < .05; **P ≤ .01; ***P ≤ .001.
Lack of PS in platelets affects global hemostasis assessment ex vivo and in vivo. (A-B) Whole blood activation of coagulation and clot polymerization determined by ROTEM analysis rotational (thromboelastography). (A) Extrinsic‐activated rotational thromboelastometry (EXTEM) assay, (B) extrinsically activated thromboelastometric test with cytochalasin D (FIBTEM). CT, coagulation time; CFT, clot formation time; α, α-angle; CFR, clot formation rate, MCF, maximum clot firmness; MCE, maximum clot elasticity. (C-D) Total thrombus-formation analysis on whole blood on collagen and TF-coated chip (AR-chip) at low (240 s−1, C) and high (600 s−1, D) shear rate. (E-F) Bleeding time (E) and blood loss (F) were measured after 2-mm tail transection in Pros1lox/loxPf4-Cre− (n = 7), Pros1lox/loxPf4-Cre+ (n = 7), and Pros1lox/− (n = 5) mice. All data are expressed as mean ± SEM. ns, not significant; *P < .05; **P ≤ .01; ***P ≤ .001.
The total thrombus-formation analysis system allows the quantitative analysis of thrombus formation, using microchips with thrombogenic surfaces (collagen, platelet chip; collagen plus TF, atheroma chip [AR]) at low and high shear rates. When whole blood flows through the analytical path of the chip, platelets adhere and aggregate on the surface of the collagen-coated capillary channels (platelet chip). Because the AR chip contains a single capillary channel coated with collagen and TF, the total thrombus-formation analysis system can also be used for the quantitative evaluation of white thrombus formation mediated by the activation of both coagulation and platelets.
Thrombus formation was comparable in Pros1lox/loxPf4-Cre− and Pros1lox/loxPf4-Cre+ blood when using platelet chips (supplemental Figure 6A-B). Differently, when using AR chips, the rate of the thrombus growth (T10-80) and the occlusion speed were higher in Pros1lox/loxPf4-Cre+ than in Pros1lox/loxPf4-Cre− blood at a low shear rate (Figure 3C), whereas no difference was observed between the 2 genotypes at a higher shear rate (Figure 3D).
Lack of PSplt shortens bleeding time and reduces blood loss after tail clipping
To assess the role of PSplt in hemostasis in vivo, a 2-mm tail clipping model was applied. Pros1lox/loxPf4-Cre+ mice displayed a significant reduction in bleeding time and blood loss compared with Pros1lox/loxPf4-Cre− mice (Figure 3E-F). Pros1lox/− mice bled less than Pros1lox/loxPf4-Cre− mice, whereas Pros1lox/− and Pros1lox/loxPf4-Cre+ mice displayed comparable bleeding time and blood loss (Figure 3E-F).
Lack of PSplt in platelets increases thrombus propensity at low but not at high shear rate
We used different thrombosis models to determine the effects of PS deficiency in platelets. In a first model, we induced thrombosis in vivo by injecting TF into the inferior vena cava.40 When using a high TF dosage (∼4.3 nM), no differences were observed between Pros1lox/loxPf4-Cre− and Pros1lox/loxPf4-Cre+ mice (Figure 4A; P > .05). However, after a low TF dose (1.1 nM), 81% of Pros1lox/loxPf4-Cre− mice survived, whereas only 50% of Pros1lox/loxPf4-Cre+ mice and 21% of Pros1lox/− mice survived (Figure 4B). Histologic analysis showed an increased number of thrombi in the lungs of Pros1lox/loxPf4-Cre+ mice that died within the 20-minute observation period compared with Pros1lox/loxPf4-Cre+ and Pros1lox/loxPf4-Cre− mice that survived the challenge, consistent with a higher thrombotic potential in Pros1lox/loxPf4-Cre+ mice (supplemental Figure 7). Fibrin and PS were detected in thrombi from Pros1lox/loxPf4-Cre+ mice that died during the challenge (supplemental Figure 7). We assumed that PS located in thrombi from Pros1lox/loxPf4-Cre+ mice originated from plasma.

Platelets PS limits thrombus propensity and affects thrombus composition. (A-C) TF-induced venous thromboembolism using high dose of recombinant TF in A (1/2 dilution of Innovin; ∼4.3 nM TF) and low dose in panels B and C (1/8 dilution of Innovin; ∼1.1 nM TF). In panel A, no differences were found between Pros1lox/loxPf4-Cre− (straight line; n = 14) and Pros1lox/loxPf4-Cre+ (dashed line, n = 14). (B) Pros1lox/loxPf4-Cre+ (dashed line; n = 14) and Pros1lox/− (dotted line, n = 14) mice showed higher mortality than Pros1lox/loxPf4-Cre− (straight line; n = 14) mice. (C) Thrombus formation in FeCl3-injured mesenteric arterioles recorded by intravital microscopy in Pros1lox/loxPf4-Cre− and Pros1lox/loxPf4-Cre+ mice, representative experiment (n = 7/genotype). Recorded occlusion time is shown in panel D. (E-G) Thrombi were collected 20 minutes after FeCl3 challenge and processed to confocal microscopy, pictures were taken close to the lesion side and on the top of the thrombus as shown in panel E. Confocal microscopy of the thrombi analyzed 20 minutes after the FeCl3 injury and stained for FXa and thrombin and CD41 (F; scale bar, 50 μM) as well as for insoluble fibrin, CD41, and CD62p (G; scale bar, 50 μM). Data are expressed as mean ± SEM. *P < .05; **P ≤ .01; ***P ≤ .001.
Platelets PS limits thrombus propensity and affects thrombus composition. (A-C) TF-induced venous thromboembolism using high dose of recombinant TF in A (1/2 dilution of Innovin; ∼4.3 nM TF) and low dose in panels B and C (1/8 dilution of Innovin; ∼1.1 nM TF). In panel A, no differences were found between Pros1lox/loxPf4-Cre− (straight line; n = 14) and Pros1lox/loxPf4-Cre+ (dashed line, n = 14). (B) Pros1lox/loxPf4-Cre+ (dashed line; n = 14) and Pros1lox/− (dotted line, n = 14) mice showed higher mortality than Pros1lox/loxPf4-Cre− (straight line; n = 14) mice. (C) Thrombus formation in FeCl3-injured mesenteric arterioles recorded by intravital microscopy in Pros1lox/loxPf4-Cre− and Pros1lox/loxPf4-Cre+ mice, representative experiment (n = 7/genotype). Recorded occlusion time is shown in panel D. (E-G) Thrombi were collected 20 minutes after FeCl3 challenge and processed to confocal microscopy, pictures were taken close to the lesion side and on the top of the thrombus as shown in panel E. Confocal microscopy of the thrombi analyzed 20 minutes after the FeCl3 injury and stained for FXa and thrombin and CD41 (F; scale bar, 50 μM) as well as for insoluble fibrin, CD41, and CD62p (G; scale bar, 50 μM). Data are expressed as mean ± SEM. *P < .05; **P ≤ .01; ***P ≤ .001.
In the second model, thrombus formation was recorded by intravital microscopy. In Pros1lox/loxPf4-Cre− mice, thrombi grew to occlusive size in about 20 minutes, and all injured arterioles were occluded (Figure 4C-D). Differently, in Pros1lox/loxPf4-Cre+ mice, occlusive thrombi were already formed in about 10 minutes, and all injured arterioles were occluded (Figure 4C-D). The formation of small emboli was observed in some Pros1lox/loxPf4-Cre− and Pros1lox/loxPf4-Cre+ mice. Notably, the size of the thrombi formed in Pros1lox/loxPf4-Cre+ mice was larger in all tested animals compared with the size of those formed in Pros1lox/loxPf4-Cre− mice, as shown by the representative images (Figure 4C). The architecture and the composition of the thrombi were studied by confocal microscopy (Figure 4E-G). More diffuse localization of FXa and thrombin was detected in thrombi from Pros1lox/loxPf4-Cre+ than from Pros1lox/loxPf4-Cre− mice (Figure 4F). Consistently, Pros1lox/loxPf4-Cre+ thrombi showed an intense fibrin staining all over the vessel, whereas fibrin staining remained localized to the injury site in Pros1lox/loxPf4-Cre− mice (Figure 4G). Activated platelets were observed close to the injury in both genotypes. In Pros1lox/loxPf4-Cre+ mice, highly activated platelets were also found at the top of the thrombus. In sharp contrast, the top of the thrombus of Pros1lox/loxPf4-Cre− mice was covered mainly by P-selectin negative platelets, indicating their less activated status (Figure 4G).
In the third model, we dynamically monitored the thrombus formation in the inferior vena cava of mice after FeCl3-injury. The thrombus volume was much larger in Pros1lox/loxPf4-Cre+ than in Pros1lox/loxPf4-Cre− mice (Figure 5A). Histology of the middle part of the thrombus (sample center, Figure 5B) revealed that the thrombus was built up in layers defined as lines of Zahn in both genotypes, but was more occlusive and richer in fibrin in Pros1lox/loxPf4-Cre+ than in Pros1lox/loxPf4-Cre− mice (Figure 5B, hematoxylin/eosin staining). Immunohistochemistry confirmed the larger amount of fibrin in the Pros1lox/loxPf4-Cre+ thrombus compared with the Pros1lox/loxPf4-Cre− thrombus (Figure 5B, fibrin staining). PS staining was detected in both genotypes, although it was more intense in Pros1lox/loxPf4-Cre− than in Pros1lox/loxPf4-Cre+ mice (Figure 5B, PS staining). Importantly, in both genotypes, PS staining was stronger in the border of the fibrin layers. Platelets were present within the whole thrombus in both genotypes (Figure 5B, CD31 staining). Section electron microscopy of the center of the thrombus revealed a higher density in the fibrin network in Pros1lox/loxPf4-Cre+ than in Pros1lox/loxPf4-Cre− mice (supplemental Figure 8A).
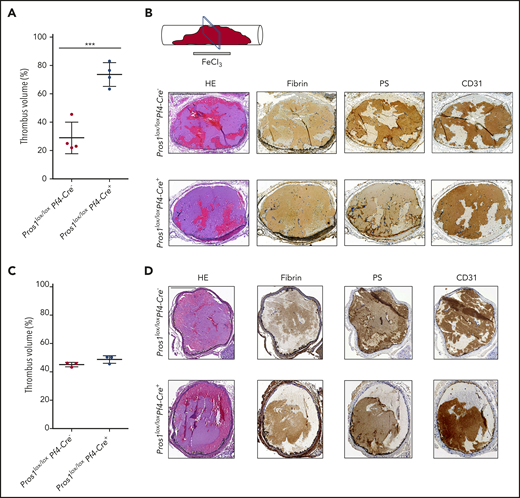
Platelets PS limits thrombosis propensity in the vena cava, but not in the carotid artery. (A) Evaluation of the thrombus volume in the vena cava 30 minutes after the FeCl3 injury, using ultra-high-frequency US (n = 4/genotype). Using the Vevo 3100 imaging system (Fujifilm VisualSonics Canada) and a MX550S linear array transducer (bandwidth, 32-55 MHz; center frequency, 40 MHz), 3-dimensional image series were acquired after 30 minutes of thrombus induction at 76-µm intervals. Using the VevoLab software package (Fujifilm VisualSonics), the 3-dimensional image series were used to make a 3-dimensional reconstruction of the inferior vena cava or carotid artery and a 3-dimensional reconstruction of the thrombus. The vessel and the thrombus area were measured by the software in cubic millimeters. The thrombus volume (%) refers to the volume (in cubic millimeters) that the thrombus occupies inside the volume (mm3) of the vessel. (B) Thrombi collected at the end of the experiment (=30 minutes after the FeCl3 injury) and processed for immunohistochemistry; the sections were performed in the middle of the vena cava thrombus, as shown. Hematoxylin and eosin staining (HE) and immunohistochemistry for insoluble fibrin, PS, and CD31 were realized in Pros1lox/loxPf4Cre− and Pros1lox/loxPf4Cre+ vena cava thrombi 30 minutes after the FeCl3 injury. Scale bar, 500 µm. (C) Evaluation of the thrombus volume in the carotid artery 30 minutes after the FeCl3 injury, using ultrahigh-frequency US (n = 3/genotype). For the method of evaluation of the thrombus volume, see panel A. (D) HE and immunohistochemistry for insoluble fibrin, PS, and CD31 were realized in Pros1lox/loxPf4Cre− and Pros1lox/loxPf4Cre+ carotid artery thrombi 30 minutes after the FeCl3 injury. Scale bar, 200 µm.
Platelets PS limits thrombosis propensity in the vena cava, but not in the carotid artery. (A) Evaluation of the thrombus volume in the vena cava 30 minutes after the FeCl3 injury, using ultra-high-frequency US (n = 4/genotype). Using the Vevo 3100 imaging system (Fujifilm VisualSonics Canada) and a MX550S linear array transducer (bandwidth, 32-55 MHz; center frequency, 40 MHz), 3-dimensional image series were acquired after 30 minutes of thrombus induction at 76-µm intervals. Using the VevoLab software package (Fujifilm VisualSonics), the 3-dimensional image series were used to make a 3-dimensional reconstruction of the inferior vena cava or carotid artery and a 3-dimensional reconstruction of the thrombus. The vessel and the thrombus area were measured by the software in cubic millimeters. The thrombus volume (%) refers to the volume (in cubic millimeters) that the thrombus occupies inside the volume (mm3) of the vessel. (B) Thrombi collected at the end of the experiment (=30 minutes after the FeCl3 injury) and processed for immunohistochemistry; the sections were performed in the middle of the vena cava thrombus, as shown. Hematoxylin and eosin staining (HE) and immunohistochemistry for insoluble fibrin, PS, and CD31 were realized in Pros1lox/loxPf4Cre− and Pros1lox/loxPf4Cre+ vena cava thrombi 30 minutes after the FeCl3 injury. Scale bar, 500 µm. (C) Evaluation of the thrombus volume in the carotid artery 30 minutes after the FeCl3 injury, using ultrahigh-frequency US (n = 3/genotype). For the method of evaluation of the thrombus volume, see panel A. (D) HE and immunohistochemistry for insoluble fibrin, PS, and CD31 were realized in Pros1lox/loxPf4Cre− and Pros1lox/loxPf4Cre+ carotid artery thrombi 30 minutes after the FeCl3 injury. Scale bar, 200 µm.
In the fourth model, thrombus formation was monitored in the carotid artery after the FeCl3-injury. Importantly, and in sharp contrast with the findings obtained with the 3 other models, the thrombus volume did not differ between Pros1lox/loxPf4-Cre− and Pros1lox/loxPf4-Cre+ mice (Figure 5C). Notably, hematoxylin/eosin, fibrin, PS, and CD31 staining did not differ between the 2 genotypes (Figure 5D). In addition, the density of the fibrin network in the center of the thrombus was comparable in Pros1lox/loxPf4-Cre− and Pros1lox/loxPf4-Cre+ mice (supplemental Figure 8B).
Effect of the lack of PSplt on clot contraction
Clot contraction upon thrombin treatment at a concentration of 1 U/mL was comparable in Pros1lox/loxPf4-Cre− and Pros1lox/loxPf4-Cre+ mice (supplemental Figure 9A), as expected in the context of a normal platelet function. In contrast, clot contraction on treatment with a higher concentration of thrombin (10 U/mL) was impaired in Pros1lox/loxPf4-Cre+ mice (Figure 6A). Indeed, clots from Pros1lox/loxPf4-Cre+ comprised a denser fibrin network than those from Pros1lox/loxPf4-Cre− mice (Figure 6B-D). Fibrin at the exterior face of the clot and platelet-rich plasma (PRP)-air interface formed a film in Pros1lox/loxPf4-Cre− mice as described in Macrae et al41 (Figure 6B). In contrast, in Pros1lox/loxPf4-Cre+, the exterior face of the clot looked porous without displaying an aspect of a sheet.41 Inside the clot, the fibrin network was more dense and compact in Pros1lox/loxPf4-Cre+ than in Pros1lox/loxPf4-Cre− mice explaining the impairment in clot contraction in Pros1lox/loxPf4-Cre+ mice (Figure 6C). Clots from Pros1lox/loxPf4-Cre+ PRP showed a denser fibrin network than did those from Pros1lox/loxPf4-Cre− PRP clots (Figure 6D).
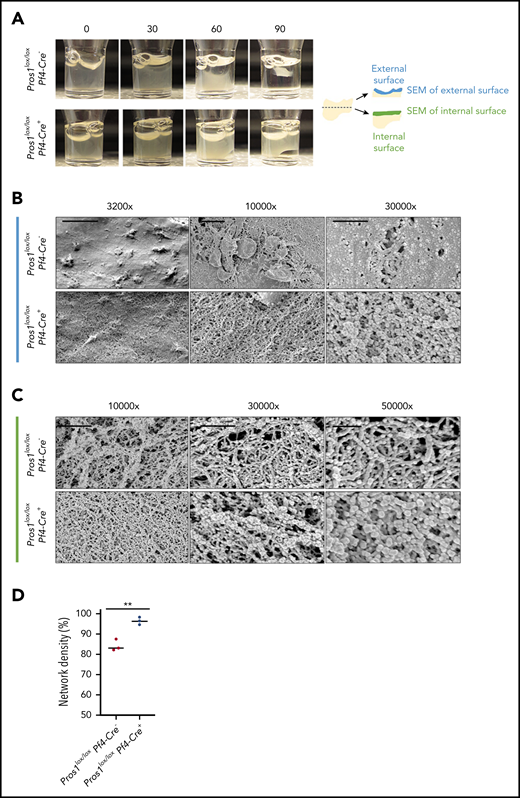
Effect of the lack of PS in platelets on clot contraction. (A) Representative images of clot contraction at different points after induction with thrombin 10 U/mL in Pros1lox/loxPf4Cre− and Pros1lox/loxPf4Cre+ PRP (n = 3/genotype). (B) Representative SEM images of the contracted clot at the external surface at different magnifications: scale bar, 10, 2, and 1 µm for ×3200, ×10 000, and ×30 000, respectively). (C) Representative SEM images of the contracted clot in the middle of the clot at different magnifications: scale bar, 2 and 1 µm for ×10 000 and ×30 000, respectively; scale bar, 500 nm for ×50 000. (D) Quantification of fibrin network density in Pros1lox/loxPf4Cre− and Pros1lox/loxPf4Cre+ mice. Measurements are presented as mean ± SEM. **P ≤ .01.
Effect of the lack of PS in platelets on clot contraction. (A) Representative images of clot contraction at different points after induction with thrombin 10 U/mL in Pros1lox/loxPf4Cre− and Pros1lox/loxPf4Cre+ PRP (n = 3/genotype). (B) Representative SEM images of the contracted clot at the external surface at different magnifications: scale bar, 10, 2, and 1 µm for ×3200, ×10 000, and ×30 000, respectively). (C) Representative SEM images of the contracted clot in the middle of the clot at different magnifications: scale bar, 2 and 1 µm for ×10 000 and ×30 000, respectively; scale bar, 500 nm for ×50 000. (D) Quantification of fibrin network density in Pros1lox/loxPf4Cre− and Pros1lox/loxPf4Cre+ mice. Measurements are presented as mean ± SEM. **P ≤ .01.
This indicates that clot retraction driven by platelet-generated contractile forces42 was normal in absence of PSplt (supplemental Figure 9A). However, in the presence of a very high concentration of thrombin, clot composition was different between the 2 genotypes, and for this reason, clot contraction did not occur to the same extent in Pros1lox/loxPf4-Cre− and Pros1lox/loxPf4-Cre+ mice (Figure 6A). Clots from Pros1lox/loxPf4-Cre+ PRP showed a denser fibrin network than did those from Pros1lox/loxPf4-Cre− PRP clots (Figure 6B-D). Similar results were obtained when PRP used for clot contraction analyses lacked PS in plasma, confirming that PSplt.is responsible for this phenotype (supplemental Figure 9B-C).
Absence of PS in platelets affects resistance to APC- and TFPI-dependent PS activity
Ex vivo TF-initiated thrombin generation assays were used to investigate the effect of the loss of PS expression in platelets on thrombin generation.
To assess whether Pros1lox/loxPf4-Cre+ mice exhibited defective functional APC-dependent PS activity, we used thrombin generation testing in Ca2+ ionophore-activated PRP in the absence of APC, in the presence of WT-rmAPC, or in the presence of rmAPC L38D (a variant with ablated PS cofactor activity).43 In this assay, APC titration showed that the addition of 6 nM of WT-rmAPC was able to reduce endogenous thrombin potential (ETP) by 90% in activated PRP of WT mice, whereas the same concentration of rmAPC L38D diminished ETP by only 25% in these mice. On the basis of these data, thrombin generation curves were recorded for activated PRP (3 mice/assay; Figure 7A-B). The calculated APC ratio (ETP+APCWT/ETP+APCL38D) indicated no APC resistance in Pros1lox/loxPf4-Cre− and Pros1lox/loxPf4-Cre+ PRP (0.19 ± 0.05 vs 0.24 ± 0.11, respectively; P > .05).
![Absence of PS expression in platelets affects resistance to APC- and TFPI-dependent PS activity. (A-B) APC-dependent activity assessed by thrombin generation in PRP (150 × 109/L platelets). Representative thrombin generation curves in presence of different amount of WT mAPC and L38D mAPC or buffer (no APC) in PRP from Pros1lox/loxPf4-Cre− (A) and Pros1lox/loxPf4-Cre+ mice (B). (C-D) Washed platelet suspension (150 G/L) from Pros1lox/loxPf4-Cre− (C) and Pros1lox/loxPf4-Cre+ mice (D) were reconstituted in platelet-free plasma (PFP) from F8−/−Pros1−/− mice, lacking plasma PS to evaluate the contribution of platelet PS to APC activity. (E-F) To evaluate the contribution of platelet PS to the thrombin potential, thrombin generation was measured in Pros1lox/loxPf4-Cre− and Pros1lox/loxPf4-Cre+ mice. ETP values from PRP (200 G/L platelets) (E) and PFP (F) are shown for both genotypes. (G) ETP change between PRP and PFP [(ETPPRPx100/ETPPFP)-100)] in Pros1lox/loxPf4-Cre− and Pros1lox/loxPf4-Cre+ mice. Data are expressed as mean ± SEM. *P < .05; **P ≤ .01. (H) FXa in activated Pros1lox/loxPf4-Cre− and Pros1lox/loxPf4-Cre+ platelets; representative experiment obtained from a pool of 5 to 6 mice/genotype.](https://ash.silverchair-cdn.com/ash/content_public/journal/blood/135/22/10.1182_blood.2019003630/4/m_bloodbld2019003630f7.png?Expires=1769096571&Signature=hxKLo~E2Q2ldlhkjTlsI1cW3Lx3FkNGt9NVpMB7WoM~qkiEwj5xuigxoirIRqcIQualu1Zipawepo8QyZKqC3WS2uQD~fizGOwof1Ly-63RB0n0Uyldjjn8OYZhjaJjwDWMNrQ048zjy4kJvBz-h32thE-qlTpTZ2PBI56yhpsiz0mn7YxzGb33yX7dIpDGY4116f6cs6n95T21sYJzpkKql7NyklYyLsahrTxvSCi6O7ur5wp89sgH6AzNnICdJBedj8Dd4DxeEDCkyDUg7EhpMnwDdG3SFvPy92RhiQAVAI26sSMuGHqYWHdxASjIIlHfIW4xZdvyQzJvrZGHbYg__&Key-Pair-Id=APKAIE5G5CRDK6RD3PGA)
Absence of PS expression in platelets affects resistance to APC- and TFPI-dependent PS activity. (A-B) APC-dependent activity assessed by thrombin generation in PRP (150 × 109/L platelets). Representative thrombin generation curves in presence of different amount of WT mAPC and L38D mAPC or buffer (no APC) in PRP from Pros1lox/loxPf4-Cre− (A) and Pros1lox/loxPf4-Cre+ mice (B). (C-D) Washed platelet suspension (150 G/L) from Pros1lox/loxPf4-Cre− (C) and Pros1lox/loxPf4-Cre+ mice (D) were reconstituted in platelet-free plasma (PFP) from F8−/−Pros1−/− mice, lacking plasma PS to evaluate the contribution of platelet PS to APC activity. (E-F) To evaluate the contribution of platelet PS to the thrombin potential, thrombin generation was measured in Pros1lox/loxPf4-Cre− and Pros1lox/loxPf4-Cre+ mice. ETP values from PRP (200 G/L platelets) (E) and PFP (F) are shown for both genotypes. (G) ETP change between PRP and PFP [(ETPPRPx100/ETPPFP)-100)] in Pros1lox/loxPf4-Cre− and Pros1lox/loxPf4-Cre+ mice. Data are expressed as mean ± SEM. *P < .05; **P ≤ .01. (H) FXa in activated Pros1lox/loxPf4-Cre− and Pros1lox/loxPf4-Cre+ platelets; representative experiment obtained from a pool of 5 to 6 mice/genotype.
Absence of PS expression in platelets affects resistance to APC- and TFPI-dependent PS activity. (A-B) APC-dependent activity assessed by thrombin generation in PRP (150 × 109/L platelets). Representative thrombin generation curves in presence of different amount of WT mAPC and L38D mAPC or buffer (no APC) in PRP from Pros1lox/loxPf4-Cre− (A) and Pros1lox/loxPf4-Cre+ mice (B). (C-D) Washed platelet suspension (150 G/L) from Pros1lox/loxPf4-Cre− (C) and Pros1lox/loxPf4-Cre+ mice (D) were reconstituted in platelet-free plasma (PFP) from F8−/−Pros1−/− mice, lacking plasma PS to evaluate the contribution of platelet PS to APC activity. (E-F) To evaluate the contribution of platelet PS to the thrombin potential, thrombin generation was measured in Pros1lox/loxPf4-Cre− and Pros1lox/loxPf4-Cre+ mice. ETP values from PRP (200 G/L platelets) (E) and PFP (F) are shown for both genotypes. (G) ETP change between PRP and PFP [(ETPPRPx100/ETPPFP)-100)] in Pros1lox/loxPf4-Cre− and Pros1lox/loxPf4-Cre+ mice. Data are expressed as mean ± SEM. *P < .05; **P ≤ .01. (H) FXa in activated Pros1lox/loxPf4-Cre− and Pros1lox/loxPf4-Cre+ platelets; representative experiment obtained from a pool of 5 to 6 mice/genotype.
To unmask the role of PSplt in thrombin generation testing in PRP, we resuspended Pros1lox/loxPf4Cre− and Pros1lox/loxPf4-Cre+ washed platelets in F8−/−Pros1−/− PFP.26 In this system, platelets were the only source of PS. Calculated APC ratio showed an APC resistance in Pros1lox/loxPf4-Cre+, but not in Pros1lox/loxPf4Cre− mice (1.16 ± 0.14 vs 0.39 ± 0.07, respectively, 3 mice/assay; P = .039; Figure 7C-D). Consistently, calculated TM ratio revealed a TM resistance in Pros1lox/loxPf4-Cre+, but not in Pros1lox/loxPf4Cre− mice (ETP ratio: 0.63 0.07 vs 0.28 0.13, respectively, 3 mice/assay [P = .013]; peak ratio: 0.76 0.01 vs 0.50 0.11, respectively, 3 mice/assay [P = .017]).
APC-dependent PS activity was also tested in PFP from Pros1lox/loxPf4-Cre+ and Pros1lox/loxPf4-Cre− mice in the presence of 8 nM WT APC and L38D APC. Calculated APC ratio showed no APC resistance in Pros1lox/loxPf4Cre+ and Pros1lox/loxPf4Cre− mice (1.40 ± 0.08 vs 1.30 ± 0.02, respectively, 2 mice/assay; P = .27), as expected because of similar PS content in PFP for both genotypes.
To evaluate the TFPI-dependent PS activity, thrombin generation was evaluated in PRP samples according to Winckers et al44 with minor modifications. To evaluate the contribution of platelet content to the thrombin generation, PRPs (200 × 109/L) from both genotypes were triggered by TF 1 pM. Both ETP and thrombin peak, the most sensitive TGA parameters for TFPI activity,44,45 were comparable in Pros1lox/loxPf4-Cre− and Pros1lox/loxPf4Cre+ mice PRP (ETP: 45132 vs 49570 nmol/L per minutes; thrombin peak: 59 ± 8 vs 68 ± 13 nM; n = 4 assay/3 mice/group; P > .05; Figure 7E; supplemental Figure 10A). ETP and thrombin peak did also not differ between Pros1lox/loxPf4-Cre− and Pros1lox/loxPf4Cre+ mice when measured in PFP (ETP: 47261 vs 34262 nmol/L per minutes; thrombin peak: 77 ± 9 vs 46 ± 8 nM; n = 4 assay/3 mice/group; P > .05; Figure 7F; supplemental Figure 10B). However, when we combined PRP thrombin generation curves with those of the corresponding PFP, we observed an inverted trend: absolute values for both ETPs were higher in PRP compared with PFP from Pros1lox/loxPf4-Cre+ mice, whereas they were lower in PRP as compared with PFP isolated from Pros1lox/loxPf4-Cre− mice. The calculated ratio between PRP and PFP (ETP change, as defined in “Materials and methods”) exhibited better this inverted relationship, suggesting that, in the presence of platelets, the thrombin potential is significantly higher in Pros1lox/loxPf4-Cre+ than in Pros1lox/loxPf4-Cre− mice (Figure 7G).
To investigate whether PSplt has a relevant effect on FXa TFPIα-driven inhibition, FXa activity was measured in a more simplified and reconstituted system. Frozen washed platelets were centrifuged to separate membranes from the intracellular content and incubated with FXa. FXa residual activity was then measured after 1 hour (Figure 7H). No FXa inhibition was promoted by the platelet membranes from both Pros1lox/loxPf4-Cre− and Pros1lox/loxPf4-Cre+ mice (data not shown). In contrast, evaluation of the remaining FXa activity in the solution phase showed that Pros1lox/loxPf4-Cre− samples promoted more efficiently the inhibition of FXa activity in comparison with Pros1lox/loxPf4-Cre+ samples. Taking into account that both genotypes do not have different concentrations of TFPI in platelets (supplemental Figure 3A), these data suggest that, in the absence of PS in platelets, TFPIα was not able to efficiently inhibit FXa activity, thus resulting in increased thrombin generation.
Discussion
Our extensive data show that PSplt controls thrombus propensity when the shear rate is low, such as in large veins, but not when shear rate is high, as in large arteries in murine thrombosis models. This concept is depicted schematically in supplemental Figure 11, where the effects of the presence (panels A,C,E) or absence (panels B,D,F) of PSplt are represented.
At a low shear rate, PSplt is secreted on platelet activation and acts as a cofactor for both APC and TFPI, thereby controlling FXa and thrombin generation within the thrombus. This dual role makes PSplt a key regulator of FXa and thrombin generation in the platelet synapse.46 Moreover, highly activated platelets and fibrin remain located at the injury boundary site. At the luminal surface, there are minimally highly activated platelets and no fibrin. The spatial heterogeneity of platelet activation and fibrin formation has been recently described on the basis of studies on thrombus formation in the jugular vein.47 One very notable finding here is that, without PSplt, the spatial organization of the thrombus is disturbed, with highly activated platelets and fibrin being distributed homogeneously in the thrombus. This is because of the diffuse activity of both FXa and thrombin within the thrombus. However, when tested in PRP, both APC and TFPI cofactor activities are preserved in the absence of PSplt because plasma PS present at normal concentrations is sufficient and there is no need of PSplt in this purified system. Our findings highlight the importance of small amounts of PS delivered by platelets within the growing thrombus for the preservation of the thrombus architecture in vivo. Moreover, at high thrombin concentrations, clot contraction does not occur in the absence of PSplt because of the too high density of the fibrin network. In addition, in absence of PSplt, the blood-air interface of the clot is not covered by a film, as expected.41 Therefore, the clot surface remains highly porous in absence of PSplt.
During thrombus formation at high shear rate, such as in large arteries, platelets accumulate faster than at low shear rate.48 For this reason, most of the platelets in the arterial thrombus do not become fully activated48 and, therefore, do not release the content of their granules, including PSplt. As a consequence, both in the presence and absence of PSplt, highly activated platelets remain confined to the site of injury, where most of the fibrin is found, indicating that thrombin generation occurs primarily if not exclusively at the injury boundary site, where TF is expressed.48 Thus, in large arteries, the size and the architecture of the thrombus do not depend on PSplt, because the release of PSplt during thrombus growth is very limited.
In summary, in presence of PSplt and at low shear rate, thromboxane A2/thromboxane receptor and ADP/P2Y12 signaling promote low-level platelet activation and adhesion in the luminal part of the thrombus (or outer shell of the thrombus), where thrombin activity is rapidly controlled.47,49 Our findings extend current knowledge by demonstrating that PSplt is a key player in the control of thrombin activity in venous, but not in arterial thrombus. They offer insights into the organization of the thrombus and highlight critical differences between arterial and venous thrombosis pathogenesis. It will be interesting to explore whether our results help to understand the effect of vitamin K antagonists as compared with other anticoagulants, especially direct oral anticoagulants. Because vitamin K antagonists not only impair plasma PS but also PSplt levels and functioning,22,50 it may be envisioned that the reduced PSplt activity in patients under vitamin K antagonists may have an effect on thrombus size and structure. In addition, PSplt might constitute a valuable therapeutic target in patients with bleeding disorders.
The online version of this article contains a data supplement.
The publication costs of this article were defrayed in part by page charge payment. Therefore, and solely to indicate this fact, this article is hereby marked “advertisement” in accordance with 18 USC section 1734.
Acknowledgments
The authors thank Haenni Beat and Helga Maria Mogel (Microscopy Imaging Center of the University of Bern) for the electron microscopy sample preparation and imaging; José A. Galván Hernández, Patricia Ney, and Irene Centos-Ramos (Translational Research Unit Platform of the University of Bern) for the microscopy sample preparation and immunohistochemistry; Denise Stalder Zeerleder (Center for Laboratory Medicine, Inselspital, Bern University Hospital) for the support for the flow cytometric analysis; Jens Stein and Neda Haghayegh (Microscopy Imaging Center of the University of Bern) for the support for the intravital microscopy experiments; David Ginsburg (University of Michigan) for providing the antibody against murine factor V; and Monica Azevedo and Beatrice Ternon for technical support.
This work was supported by the Swiss National Foundation for Scientific Research grants PPOOB-106690/1, PPOOP3-123430, 310030_153436, 314730_173127, and 316030_177126; the Pierre Mercier pour la Science Foundation; the CSL‐Behring‐Prof. Heimburger Award; the Bayer Hemophilia Award; La Fondation Dinu Lipatti–Dr Henri Dubois-Ferrière; the Novartis Foundation for Medical-Biological Research (A.A.-S.); and National Institutes of Health, National Heart, Lung, and Blood Institute grants RO1 HL133728 and HL142975 (J.H.G.).
Authorship
Contribution: S.C. made the figures; S.C., R.P.-E., F.S., L. Bologna, A.C.B., C.Q., M.D.R.C., V.E., T.M.H., and A.A.-S. performed research and/or analyzed the data; S.C., R.P.-E., and A.A.-S. designed experiments and wrote the paper; L. Burnier, Y.M., J.A.F., and J.H.G. provided reagents and discussed results; A.A.-S. conceptualized the study; and all authors discussed the results and commented on the manuscript.
Conflict-of-interest disclosure: The authors declare no competing financial interests.
Correspondence: Anne Angelillo-Scherrer, Department of Hematology and Central Hematology Laboratory, Inselspital, Bern University Hospital, University of Bern, CH-3010 Bern, Switzerland; e-mail: anne.angelillo-scherrer@insel.ch.

This feature is available to Subscribers Only
Sign In or Create an Account Close Modal