

Key Points
EPCR deficiency protects against hemophilic joint disease by reducing joint bleeding.
Administration of a single dose of EPCR-blocking antibody attenuates the progression of hemophilic arthropathy in hemophilia A mouse model.

Abstract
We recently showed that clotting factor VIIa (FVIIa) binding to endothelial cell protein C receptor (EPCR) induces anti-inflammatory signaling and protects vascular barrier integrity. Inflammation and vascular permeability are thought to be major contributors to the development of hemophilic arthropathy following hemarthrosis. The present study was designed to investigate the potential influence of FVIIa interaction with EPCR in the pathogenesis of hemophilic arthropathy and its treatment with recombinant FVIIa (rFVIIa). For this, we first generated hemophilia A (FVIII−/−) mice lacking EPCR (EPCR−/−FVIII−/−) or overexpressing EPCR (EPCR++ FVIII−/−). Joint bleeding was induced in FVIII−/−, EPCR−/−FVIII−/−, and EPCR++FVIII−/− mice by needle puncture injury. Hemophilic synovitis was evaluated by monitoring joint bleeding, change in joint diameter, and histopathological analysis of joint tissue sections. EPCR deficiency in FVIII−/− mice significantly reduced the severity of hemophilic synovitis. EPCR deficiency attenuated the elaboration of interleukin-6, infiltration of macrophages, and neoangiogenesis in the synovium following hemarthrosis. A single dose of rFVIIa was sufficient to fully prevent the development of milder hemophilic synovitis in EPCR−/−FVIII−/− mice. The development of hemophilic arthropathy in EPCR-overexpressing FVIII−/− mice did not significantly differ from that of FVIII−/− mice, and 3 doses of rFVIIa partly protected against hemophilic synovitis in these mice. Consistent with the data that EPCR deficiency protects against developing hemophilic arthropathy, administration of a single dose of EPCR-blocking monoclonal antibodies markedly reduced hemophilic synovitis in FVIII−/− mice subjected to joint bleeding. The present data indicate that EPCR could be an attractive new target to prevent joint damage in hemophilia patients.
Introduction
Frequent joint bleeding in hemophilia patients results in hemophilic arthropathy (HA), a debilitating, degenerative joint disease with a significant negative impact on mobility and quality of life.1-3 HA typically begins synovitis that is characterized by synovial hyperplasia, migration of inflammatory cells, and a high degree of neoangiogenesis in the synovium, followed by the destruction of articular cartilage and subchondral bone.4-7 Iron deposition in the synovium from repeated joint bleeding is thought to play a crucial role in the pathogenesis of HA. Iron was shown to cause toxicity for articular chondrocytes,8 upregulate critical genes such as c-myc and mdm2 that promote the proliferation of synovial fibroblasts,9,10 and induce the expression of several proinflammatory cytokines.5 Blood-derived mononuclear cells and subsequently activated synoviocytes and chondrocytes were also shown to produce proinflammatory cytokines in the affected joint.11 Joint bleed–induced inflammatory cytokines in the synovium, particularly interleukin-1β (IL-1β), tumor necrosis factor α (TNF-α), and IL-6, appear to play a leading role in the pathogenesis of HA, as they could elicit synovial hyperplasia, increase vascular permeability, activate matrix metalloproteases, induce apoptosis of chondrocytes, and destruction of cartilage and bone.11,12 Consistent with a potential key role for inflammation in the pathogenesis of HA, recent studies showed that blood-induced joint damage and bone loss could be prevented by blocking IL-1β by monoclonal antibody (mAb) or receptor agonist13 or blocking the iRhom2/ADAM17/TNF-α pathway with inactivation of iRhom2 or TNF-α or anti-TNF-α (etanercept).14 Supporting the concept that blocking inflammation could provide protective effect in HA, Narkbunnam et al15 reported that the administration of anti–IL-6R with factor VIII (FVIII) replacement protected hemophilia A mice more effectively against bleeding-induced arthropathy.
Hemophilic joint bleeding, in addition to eliciting inflammation, also leads to the elevation in vascular permeability16,17 and neoangiogenesis.18 Acharya et al18 showed the presence of potent proangiogenic mediators, including vascular endothelial growth factor (VEGF), hematopoietic, and endothelial progenitor cells in the synovium of patients with hemophilic joint disease. Increased vascular permeability and remodeling associated with hemarthrosis may promote rebleeding events that accelerate the progression of HA.17
At present, efforts to prevent HA are primarily focused on the management of acute bleeds and optimizing the dose and schedule for prophylactic factor replacement.19,20 Although factor replacement therapy limits the incidence of joint bleeds, HA cannot be avoided completely with clotting factor replacement even on the best prophylaxis protocols, as breakthrough bleeds can occur in these patients.21-23 Furthermore, manifestation and severity of HA vary between hemophilia patients, indicating that the response to bleeds can differ across patients.6 Therefore, in addition to factor replacement, disease-modifying treatments, such as anti-inflammatory therapy, may hold promise in treating HA.13-15,24
Recombinant FVIIa (rFVIIa) has been used widely for >2 decades to treat bleeding disorders in hemophilia patients with inhibitors and other groups of patients.25-27 A recent review of the literature provides strong evidence that prophylaxis with rFVIIa is effective in reducing target joint bleeds in hemophilia patients with inhibitors.28 We29 and others30,31 showed that FVIIa binds endothelial cell protein C receptor (EPCR), a key protein in the activated protein C (APC)–mediated anticoagulant pathway.32 Our recent studies showed that pharmacological concentrations of rFVIIa downregulate the EPCR-mediated anticoagulant pathway by displacing protein C from the EPCR, which contributes to the hemostatic effect of rFVIIa in hemophilia treatment.33 In additional studies, we found that FVIIa binding to EPCR induces PAR1-mediated cell signaling.34 FVII-EPCR-PAR1–induced signaling was shown to protect against VEGF-induced barrier permeability35 and cytokine- and lipopolysaccharide (LPS)-induced inflammation.36 Since inflammation and vascular permeability play pivotal roles in the pathogenesis of HA, we hypothesized that EPCR-FVIIa–induced anti-inflammatory effects and barrier stabilization would be beneficial in treating HA. The present study aimed to investigate the role of EPCR in the pathophysiology of HA and its influence on rFVIIa treatment of the disease.
The data presented here show that EPCR deficiency protects against HA in an experimental murine HA model system. Furthermore, our data reveal that blocking EPCR in FVIII−/− mice with a blocking mAb strikingly reduced synovial inflammation, macrophage infiltration, and neoangiogenesis that develop in the mouse following needle puncture-induced joint bleeding. Our studies identify EPCR as a potential target for the therapy of HA.
Materials and methods
Additional details can be found in supplemental Materials and methods (available on the Blood Web site).
Reagents
Human rFVIIa was provided the late Walter Kisiel, University of New Mexico, Albuquerque, NM. Monoclonal antibodies against mouse EPCR were prepared by immunizing rats with recombinant mouse soluble EPCR.37
Animals
EPCR−/−FVIII−/− mice were generated first crossing Procrflox/flox and Procr+/floxMeox2+/cre mice with FVIII−/− mice to generate Procrflox/floxFVIII−/− and Procr+/floxMeox2+/creFVIII−/− mice, and then crossing female Procrflox/floxFVIII−/− with male Procr+/floxMeox2+/creFVIII−/− mice. EPCR+/+FVIII−/− mice were generated by crossing Tie2-EPCR mice with FVIII−/− mice.
Induction of joint bleeding by needle puncture
Intraarticular bleeding into knee joints was induced by a needle puncture injury, as described earlier.38,39 Briefly, the right knee joint capsule of anesthetized mice was punctured with a 30 × 0.5-G needle below the patella to induce bleeding in the joint. At specified times following the injury, mice were killed, the skin was removed from over the knee joints, and the knee joints were photographed and processed for histology, immunohistochemistry, or collecting synovial fluid.
Treatments
Mice were treated with either a single dose (20 minutes following the injury) or 3 doses (20 minutes, 1 and 3 days following the injury) of rFVIIa (1 mg/kg body weight) by IV injection via the tail vein. In the case of EPCR antibody treatment, a single dose of EPCR antibodies (1 mg/kg body weight) was given intraperitoneally 1 day before the injury.
Evaluation of hemarthrosis
Knee diameter, before the injury and alternate days for 2 weeks following the injury, was measured using electronic calipers, and the percent change in joint diameter was calculated. Gross examination of knee joints was performed every alternate day to assess visually the extent of blood leaked into joints and knee mobility. A visual bleeding score (VBS) was assigned to score the extent of injury (0, normal knee and absence of blood; 1, normal knee, presence of blood; 2, distended but not a tense knee, presence of blood; 3, tense and distended knee, presence of blood). The joint evaluation and the assignment of VBS were performed in a blinded fashion where the evaluator was not aware of the mouse genotype or experimental treatments.
Histology of knee joints and synovitis scoring
Joint tissue sections were stained with hematoxylin and eosin (H&E), viewed under a microscope, and scored for hemophilic synovitis as described earlier by Valentino and Hakobyan.40 Sections were also stained with Alcian blue or Safranin O/fast green to stain for proteoglycans and glycosaminoglycans to assess cartilage degeneration and Prussian blue stain to detect iron.
Immunohistochemistry
Knee joint tissue sections were stained for F4/80 antigen to evaluate the infiltration of macrophages, and CD31 to assess neoangiogenesis. The staining pattern of histology and immunohistochemistry sections were evaluated and scored in a blinded manner by an evaluator who was not aware of the experimental groups, in addition to the first author, independently. The scores were well matched.
Data analysis
Statistical significance among the groups was analyzed by either 2-way analysis of variance, repeated measures followed by Tukey’s post hoc multiple comparison test, or 1-way analysis of variance followed by Dunnett’s post hoc multiple comparison test as appropriate. When comparing 2 groups, the Mann-Whitney test was used to determine statistical significance. Survival curves were plotted using the Kaplan-Meier method, and statistical significance was determined by the log-rank (Mantel-Cox) test.
Results
EPCR deficiency protects against mortality following needle puncture–induced joint bleeding in a murine model system
Needle puncture injury was shown to induce bleeding into the joints of hemophilic mice and leads to clinical and pathological changes in joints that closely mirror human hemophilic synovitis.38 Therefore, in the present study, we adopted the above method to investigate the influence of EPCR on the pathogenesis of HA and FVIIa treatment of the disease by employing hemophilia A (FVIII−/−), EPCR-deficient FVIII−/− (EPCR−/−FVIII−/−), or EPCR-overexpressing FVIII−/− (EPCR++FVIII−/−) mice. The needle puncture injury resulted in massive joint bleeding in all 3 groups of hemophilia mice (supplemental Figure 1A). In addition to bleeding in the joint, the needle puncture also resulted in massive bleeding in the soft tissues. The bleeding pattern was consistent with the description of needle puncture induced bleeding in FVIII−/− mice in an earlier report.38 Measurement of hemoglobin levels extracted from the joint and adjacent soft tissue 5 hours following the injury showed an equal amount of joint bleeding in all 3 groups of hemophilia mice (supplemental Figure 1B). Measurement of hematocrit and hemoglobin content in peripheral blood at 5 hours following the needle injury showed a marked decrease in their levels following the injury in all 3 groups of mice, and the extent of decrease in hematocrit or hemoglobin levels was not statistically significant among the 3 groups (supplemental Figure 1C-D). The mortality rate in EPCR−/−FVIII−/− mice following the injury was significantly lower compared with FVIII−/− and EPCR++FVIII−/− mice (P < .01; supplemental Figure 2A). Approximately 50% of FVIII−/− mice and 40% of EPCR++FVIII−/− mice died within 3 days of the injury, whereas <15% of EPCR−/−FVIII−/− mice died during the same period following the injury. No deaths were observed in wild-type (WT) mice subjected to the injury. Administration of either a single dose or 3 doses of rFVIIa significantly reduced the mortality of FVIII−/− mice (supplemental Figure 2B; P < .001). Three doses of rFVIIa were required to substantially reduce the mortality of EPCR++FVIII−/− mice (supplemental Figure 2C). The minimal death rate in EPCR−/−FVIII−/− mice was further reduced by the administration of 3 doses of rFVIIa (supplemental Figure 2D).
EPCR deficiency lessens hemophilic synovitis following joint bleeding
Joint bleeding in mice was assessed for 14 days by visually inspecting mice on alternate days and assigning a VBS. Needle puncture injury in WT mice resulted in either no bleeding or barely detectable bleeding (VBS of 0-1). The VBS was significantly higher in all groups of hemophilia mice compared with WT mice (P < .0001), and no statistically significant differences were observed in the VBS among FVIII−/−, EPCR++FVIII−/−, and EPCR−/−FVIII−/− mice at day 2 (Figure 1A). However, the VBS has decreased progressively in EPCR−/−FVIII−/− mice, reaching 0 at day 12, whereas the visual bleeding was remained persistent in FVIII−/− and EPCR++FVIII−/− mice throughout the 14-day observational period (Figure 1A). Analysis of iron deposition in the synovium by Prussian blue staining as an indicator of recurrent joint bleeding showed no detectable or only traces of iron deposition in EPCR−/−FVIII−/− mice, whereas iron deposition was prominent in FVIII−/− and EPCR++FVIII−/− mice (supplemental Figure 3). The staining appears to be more intense in EPCR++FVIII−/− mice. Administration of a single dose of rFVIIa had a minimal effect on the VBS in FVIII−/− and EPCR++FVIII−/− mice, whereas administration of 3 doses of rFVIIa reduced the VBS significantly, particularly in EPCR++FVIII−/− mice (FVIII−/− mice, P < .05; EPCR++FVIII−/− mice, P < .0001; Figure 1C-E). Both 1 and 3 doses of rFVIIa markedly reduced the VBS in EPCR−/−FVIII−/− mice (Figure 1G).

Effect of EPCR deficiency and rFVIIa treatment on joint bleeding and joint edema in hemophilia A mice following needle puncture of joint. Joint bleeding was initiated by needle puncture injury. Joint bleeding was evaluated by physical examination of knee joints and assigning an arbitrary score (A,C,E,G). Knee joint diameter, before the injury and alternate days for 2 weeks following the injury, was measured using electronic calipers. The diameter of the knee joint before the injury was subtracted from the diameter following the injury, and the differences in the diameter were plotted as the percentage (B,D,F,H). (A and B) WT (green), FVIII−/− (red), EPCR++FVIII−/− (blue), and EPCR−/−FVIII−/− (magenta) mice following needle puncture of joints. (C-D) FVIII−/− mice that were left untreated (red) or treated with a single dose (at 20 minutes following needle puncture, blue) or 3 doses (at 20 minutes, day 1 and day 3 following needle puncture, green) of rFVIIa (1 mg/kg). (E-F) EPCR++FVIII−/− mice that were left untreated (red) or treated with a single dose (blue) or 3 doses (green) of rFVIIa following needle puncture-induced joint bleeding. (G-H) EPCR−/−FVIII−/− mice that were left untreated (red) or treated with a single dose (blue) or 3 doses (green) of rFVIIa following needle puncture injury. Data are plotted as mean ± standard error of the mean (SEM) (n = 6). The data were analyzed by 2-way analysis of variance with Tukey’s multiple comparison test. Statistically significant differences identified in panels A and B were between FVIII−/− and EPCR−/−FVIII−/− mice. In other panels, statistical significance was determined between untreated and rFVIIa-treated groups. *P < .05; **P < .01; ***P < .001; ****P < .0001.
Effect of EPCR deficiency and rFVIIa treatment on joint bleeding and joint edema in hemophilia A mice following needle puncture of joint. Joint bleeding was initiated by needle puncture injury. Joint bleeding was evaluated by physical examination of knee joints and assigning an arbitrary score (A,C,E,G). Knee joint diameter, before the injury and alternate days for 2 weeks following the injury, was measured using electronic calipers. The diameter of the knee joint before the injury was subtracted from the diameter following the injury, and the differences in the diameter were plotted as the percentage (B,D,F,H). (A and B) WT (green), FVIII−/− (red), EPCR++FVIII−/− (blue), and EPCR−/−FVIII−/− (magenta) mice following needle puncture of joints. (C-D) FVIII−/− mice that were left untreated (red) or treated with a single dose (at 20 minutes following needle puncture, blue) or 3 doses (at 20 minutes, day 1 and day 3 following needle puncture, green) of rFVIIa (1 mg/kg). (E-F) EPCR++FVIII−/− mice that were left untreated (red) or treated with a single dose (blue) or 3 doses (green) of rFVIIa following needle puncture-induced joint bleeding. (G-H) EPCR−/−FVIII−/− mice that were left untreated (red) or treated with a single dose (blue) or 3 doses (green) of rFVIIa following needle puncture injury. Data are plotted as mean ± standard error of the mean (SEM) (n = 6). The data were analyzed by 2-way analysis of variance with Tukey’s multiple comparison test. Statistically significant differences identified in panels A and B were between FVIII−/− and EPCR−/−FVIII−/− mice. In other panels, statistical significance was determined between untreated and rFVIIa-treated groups. *P < .05; **P < .01; ***P < .001; ****P < .0001.
In WT mice, the injury increased joint diameter minimally (<10% on day 2), whereas the joint diameter was increased by ∼55% to 70% in all 3 groups of hemophilia mice (Figure 1B). However, the joint diameter of EPCR−/−FVIII−/− mice were decreased progressively after 2 days following the injury, and the decrease was statistically significant compared with a minor decrease observed in joint diameters of FVIII−/− and EPCR++FVIII−/− mice (P < .01; Figure 1B). As with the VBS, a single dose of rFVIIa had no significant effect on the increased joint diameter of FVIII−/− mice associated with the injury, whereas 3 doses of rFVIIa showed a substantial decrease in joint diameter (Figure 1D). However, the difference did not reach statistical significance. Similar to FVIII−/− mice, a single dose of rFVIIa had no significant effect on the increased joint diameter of EPCR++FVIII−/− mice following the injury (Figure 1F). Although 3 doses of rFVIIa treatment significantly reduced the increased joint diameter of EPCR++FVIII−/− mice in the early phase, no significant differences were found in the joint diameter at day 14 among EPCR++FVIII−/− mice that received no treatment, a single dose of rFVIIa, or 3 doses of rFVIIa (Figure 1F). These data deviate from the VBS, where we found 3 doses of rFVIIa reduced the score significantly, returning to close to baseline (Figure 1E). In the case of EPCR−/−FVIII−/− mice, both a single dose and 3 doses of rFVIIa markedly reduced the joint diameter of injured EPCR−/−FVIII−/− mice (Figure 1H; P < .001). Even in the absence of treatment, the joint diameter of EPCR−/−FVIII−/− mice was significantly decreased, but unlike VBS (Figure 1G), it did not return to the baseline at day 14 (Figure 1H).
The histological examination of injured joints 14 days after the injury showed hypercellularity in the joint space of untreated FVIII−/− and EPCR++FVIII−/− mice. The joint space was filled with multiple layers of proliferating synovial fibroblasts, which led to the expansion of the synovial and stromal linings and the formation of synovial villi (Figure 2A). The synovium was filled with infiltration of macrophages (Figure 2C) and a high density of new blood vessels (Figure 3A) (for enlarged images, see supplemental Figures 4 and 5). A single dose of rFVIIa had a negligible effect on the synovial hyperplasia, macrophage infiltration, or neoangiogenesis observed in the joint space of injured knees of FVIII−/− and EPCR++FVIII−/− mice. However, 3 doses of rFVIIa significantly reduced the synovial hyperplasia (Figure 2A-B), infiltration of macrophages (Figure 2C-D), and neoangiogenesis (Figure 3A-B) in the above groups of mice. In contrast to FVIII−/− and EPCR++FVIII−/− mice, only modest changes were noted in the synovial histology in injured EPCR−/−FVIII−/− mice (Figure 2A-B). In these mice, macrophage infiltration was markedly lower (Figure 2C-D), and the neoangiogenesis was close to absent (Figure 3A). Administration of a single dose of rFVIIa fully corrected the remodeling of the synovium in EPCR−/−FVIII−/− mice (Figures 2A,C and 3A). Analysis of joint tissue sections with Alcian blue staining for the proteoglycans showed complete degeneration of cartilage in the injured joints of FVIII−/− and EPCR++FVIII−/− mice (Figure 3C). A single dose of rFVIIa partly protected the destruction of the cartilage in these mice. However, because of the variation in data, it is not statistically significant. Three doses of rFVIIa significantly protected FVIII−/− and EPCR++FVIII−/− mice from degeneration of cartilage induced by the joint bleed (Figure 3C-D). EPCR−/−FVIII−/− mice were partly protected, even in the absence of any treatment, from the degeneration of cartilage induced by bleeding into joints (Figure 3C-D). A single dose of rFVIIa was enough to fully protect EPCR−/−FVIII−/− mice from cartilage degeneration (Figure 3C-D). Staining of joint tissue sections with Safranin O/fast green stain, which also stains proteoglycans and glycosaminoglycans, confirmed the data obtained with Alcian blue staining (supplemental Figure 6).
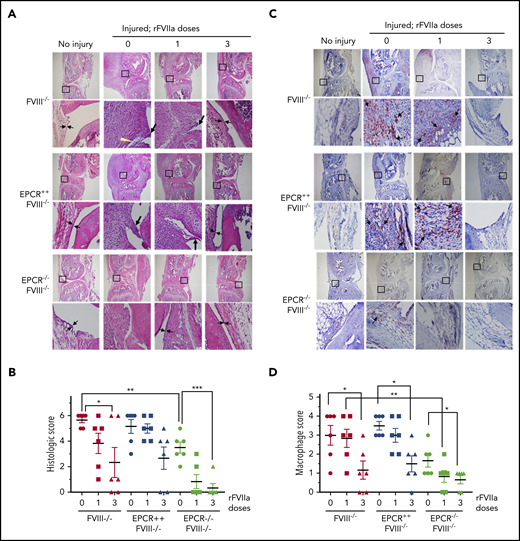
Histopathological analysis and evaluation of macrophage infiltration in knee joints of FVIII−/− , EPCR++ FVIII−/− , and EPCR−/− FVIII−/− mice 2 weeks after needle injury. Joint bleeding in FVIII−/−, EPCR++FVIII−/−, and EPCR−/−FVIII−/− mice was induced by needle puncture of joint. The mice were left either untreated (0) or treated with a single dose or 3 doses of rFVIIa, as described in the Figure 1 legend. Fourteen days after injury, knee joints were excised and sectioned, and tissue sections were stained with H&E (A) or the macrophage marker F4/80 (C). As a control, knee joint tissue sections of uninjured mice were also stained. The original magnification of images shown in the top lane is ×4. The area identified in the square box was reimaged at higher magnification (×40) and shown in the bottom lane. The histology of uninjured knee joints exhibits distal femur and proximal tibia, synovium with ≤4 cell layers of the synovial lining, and subsynovial cells associated with fat cells in the synovium with the meniscus in the center. (A) Two thin black arrows pointing each other indicate width of synovial lining layer, the yellow arrowhead points out red blood cells, and the thick black arrows point out synovial villi. (C) Arrows point out macrophages. (B) The joint tissue pathology was quantified by scoring H&E-stained sections on a 0 to 6 scale for synovial hyperplasia (0, normal, less than 4 cell layers thick; 1, 4-5 layers thick 1; 6-7 layers thick; 3, >7 layers), presence of red blood cells (0, absent; 1, present), villus formation (0, absent; 1, present), and discoloration by hemosiderin (0, absent; 1, present). (D) Macrophage infiltration was quantified on a 0 to 4 scale (0, absence of macrophages; 1, scattered macrophages; 2, line of macrophages; 3, a cluster of macrophages; 4, sheets of macrophages). *P < .05; **P < .01; ***P < .001.
Histopathological analysis and evaluation of macrophage infiltration in knee joints of FVIII−/− , EPCR++ FVIII−/− , and EPCR−/− FVIII−/− mice 2 weeks after needle injury. Joint bleeding in FVIII−/−, EPCR++FVIII−/−, and EPCR−/−FVIII−/− mice was induced by needle puncture of joint. The mice were left either untreated (0) or treated with a single dose or 3 doses of rFVIIa, as described in the Figure 1 legend. Fourteen days after injury, knee joints were excised and sectioned, and tissue sections were stained with H&E (A) or the macrophage marker F4/80 (C). As a control, knee joint tissue sections of uninjured mice were also stained. The original magnification of images shown in the top lane is ×4. The area identified in the square box was reimaged at higher magnification (×40) and shown in the bottom lane. The histology of uninjured knee joints exhibits distal femur and proximal tibia, synovium with ≤4 cell layers of the synovial lining, and subsynovial cells associated with fat cells in the synovium with the meniscus in the center. (A) Two thin black arrows pointing each other indicate width of synovial lining layer, the yellow arrowhead points out red blood cells, and the thick black arrows point out synovial villi. (C) Arrows point out macrophages. (B) The joint tissue pathology was quantified by scoring H&E-stained sections on a 0 to 6 scale for synovial hyperplasia (0, normal, less than 4 cell layers thick; 1, 4-5 layers thick 1; 6-7 layers thick; 3, >7 layers), presence of red blood cells (0, absent; 1, present), villus formation (0, absent; 1, present), and discoloration by hemosiderin (0, absent; 1, present). (D) Macrophage infiltration was quantified on a 0 to 4 scale (0, absence of macrophages; 1, scattered macrophages; 2, line of macrophages; 3, a cluster of macrophages; 4, sheets of macrophages). *P < .05; **P < .01; ***P < .001.

Evaluation of neoangiogenesis and cartilage degeneration in knee joints of FVIII−/− , EPCR++ FVIII−/− , and EPCR−/− FVIII−/− mice 2 weeks after needle puncture–induced joint bleeding. Joint bleeding in FVIII−/−, EPCR++FVIII−/−, and EPCR−/−FVIII−/− mice was induced by needle puncture of joints, and the mice were treated with a single dose or 3 doses of rFVIIa or left untreated as described in the Figure 1 legend. Fourteen days after injury, knee joints were excised, sectioned, and tissue sections were immunostained with the endothelial cell–specific marker CD31 (A) or Alcian blue for proteoglycans (C). As a control, knee joint tissue sections of uninjured mice were also stained. The original magnification of images shown in the top lane is ×4. The area identified in the square box was reimaged at higher magnification (×40) and shown in the bottom lane. Arrows point out newly formed blood vessels (A) and articular cartilage (C). (B) To quantify neoangiogenesis, the number of blood vessels positively stained with CD 31 was counted in multiple fields and averaged per field. (D) Cartilage degeneration was scored on a 0 to 2 scale (0, absence of cartilage degeneration; 1, partial loss of proteoglycan content and pannus formation; 2, complete cartilage degeneration/absence of proteoglycans, pannus formation, and femur remodeling). *P < .05; ***P < .001; ****P < .0001.
Evaluation of neoangiogenesis and cartilage degeneration in knee joints of FVIII−/− , EPCR++ FVIII−/− , and EPCR−/− FVIII−/− mice 2 weeks after needle puncture–induced joint bleeding. Joint bleeding in FVIII−/−, EPCR++FVIII−/−, and EPCR−/−FVIII−/− mice was induced by needle puncture of joints, and the mice were treated with a single dose or 3 doses of rFVIIa or left untreated as described in the Figure 1 legend. Fourteen days after injury, knee joints were excised, sectioned, and tissue sections were immunostained with the endothelial cell–specific marker CD31 (A) or Alcian blue for proteoglycans (C). As a control, knee joint tissue sections of uninjured mice were also stained. The original magnification of images shown in the top lane is ×4. The area identified in the square box was reimaged at higher magnification (×40) and shown in the bottom lane. Arrows point out newly formed blood vessels (A) and articular cartilage (C). (B) To quantify neoangiogenesis, the number of blood vessels positively stained with CD 31 was counted in multiple fields and averaged per field. (D) Cartilage degeneration was scored on a 0 to 2 scale (0, absence of cartilage degeneration; 1, partial loss of proteoglycan content and pannus formation; 2, complete cartilage degeneration/absence of proteoglycans, pannus formation, and femur remodeling). *P < .05; ***P < .001; ****P < .0001.
The global scoring of the synovitis pathology, based on the scoring system proposed by Valentino and colleagues (see supplemental Table 1), clearly showed that synovitis was less severe in EPCR−/−FVIII−/− mice compared with FVIII−/− and EPCR++FVIII−/− mice (Figure 4A). Administration of either a single dose or 3 doses of rFVIIa completely prevented the evolution of pathological changes following the exposure of the joint to blood in these mice. In FVIII−/− and EPCR++FVIII−/− mice, a single dose of rFVIIa had only a minimal effect on the synovitis score, whereas 3 doses of rFVIIa reduced the score significantly (P < .001).
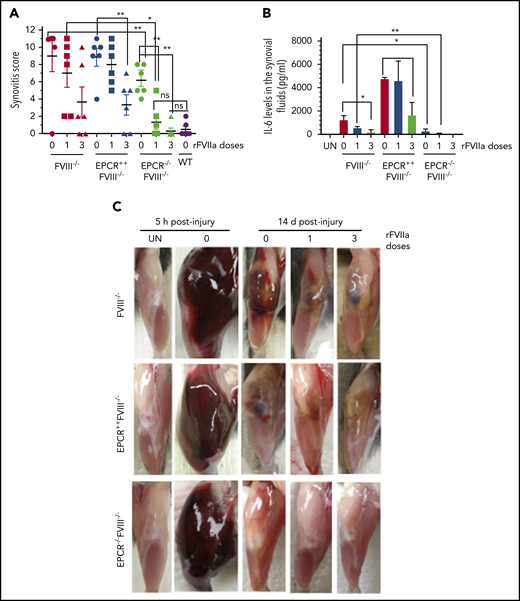
EPCR deficiency attenuates synovitis following hemarthrosis in FVIII−/− mouse joint. Joint bleeding in WT, FVIII−/−, EPCR++FVIII−/−, and EPCR−/−FVIII−/− mice was induced by needle injury, and the mice were treated with a single or 3 doses of rFVIIa or left untreated as described in the Figure 1 legend. (A) Synovitis score was determined as described in "Materials and methods," and a score of 11 was maximum. (B) IL-6 levels in synovial fluids. (C) Representative images of uninjured and injured knees. *P < .05; **P < .01; ns, no statistically significant difference.
EPCR deficiency attenuates synovitis following hemarthrosis in FVIII−/− mouse joint. Joint bleeding in WT, FVIII−/−, EPCR++FVIII−/−, and EPCR−/−FVIII−/− mice was induced by needle injury, and the mice were treated with a single or 3 doses of rFVIIa or left untreated as described in the Figure 1 legend. (A) Synovitis score was determined as described in "Materials and methods," and a score of 11 was maximum. (B) IL-6 levels in synovial fluids. (C) Representative images of uninjured and injured knees. *P < .05; **P < .01; ns, no statistically significant difference.
Measurement of inflammatory mediators in the synovial fluid showed a marked increase in IL-6 levels in the synovium of injured knees of FVIII−/− mice, and the administration of rFVIIa, particularly 3 doses of rFVIIa, reduced IL-6 levels (Figure 4B). IL-6 levels were significantly higher in the synovial fluid of injured knees of EPCR++FVIII−/− mice. One dose of rFVIIa treatment did not affect the increased IL-6 levels in these mice, whereas 3 doses of rFVIIa significantly reduced IL-6 levels. Still, IL-6 levels in rFVIIa-treated EPCR++FVIII−/− mice were similar to those measured in untreated FVIII−/− mice (Figure 4B). Interestingly, IL-6 levels were barely increased in the synovium of injured knees of EPCR−/−FVIII−/− mice, and a single dose of rFVIIa completely inhibited the elaboration of IL-6 in this group of mice (Figure 4B). Measurement of other cytokines, such as TNF-α and IL-1β, in the synovial fluid showed that their levels were below the detection limit of the assay.
Gross examination of knee joints 5 hours following the injury showed that needle puncture resulted in massive bleeding in all 3 genotypes of hemophilia mice (Figure 4C). The bleeding was observed not only in the joint but also in the soft tissues. The hindlimb of the injured joint was swollen massively, and the joint capsule was distended. The mice showed a reduced range of motion. After 14 days, the massive swelling of the joint was mostly resolved, but a significant amount of blood was still seen in the joint space of FVIII−/− and EPCR++FVIII−/− mice. In EPCR−/−FVIII−/− mice, even in the absence of rFVIIa treatment, the blood in the articular space is mostly resolved at 14 days following the injury. The injured limb of EPCR−/−FVIII−/− mice treated with rFVIIa appeared to be very similar to the uninjured limb (Figure 4C).
A single dose of EPCR-blocking mAb reduces HA following hemarthrosis
Since the above data suggest that EPCR deficiency could protect against the development of HA, next, we investigated whether blocking the EPCR function with an antibody could provide similar protection against hemophilic arthropathy. FVIII−/− mice were treated with a single dose of either EPCR-blocking mAb or nonblocking mAb, and 24 hours later, joint bleeding was induced with a needle puncture. The semiquantitative determination of joint bleeding by visual inspection of joints revealed that the administration of EPCR-blocking mAb markedly reduced the VBS, close to zero at the end of the 14-day observation period (Figure 5A). Measurement of the knee joint diameter showed that the administration of EPCR-blocking mAb curtailed the swelling of the injured knee (Figure 5B). The swelling is completely resolved at the end of the 14-day observational period. Gross examination of knee joints 14 days following the injury showed no noticeable differences between the uninjured left knee and injured right knee in mice administered with EPCR-blocking antibodies (Figure 5C).

A single dose of EPCR-blocking mAb attenuates joint bleeding and swelling in hemophilia A mice injured with a needle puncture. FVIII−/− mice were administered with either EPCR-blocking (green circles) or EPCR-nonblocking mAb (red squares) (1 mg/kg). Twenty-four hours later, joint bleeding was induced with a needle puncture. Mice were monitored for 14 days to evaluate VBS (A) and changes in joint diameter (B). At the end of 14 days, mice were killed, and the limbs were photographed after removing overlying skin (C). UN, uninjured; I, injured. Data are mean ± SEM (n = 10 to 11 mice/group) (A-B). *P < .05; **P < 0.01; ***P < .001. (C) Pictures are representative images.
A single dose of EPCR-blocking mAb attenuates joint bleeding and swelling in hemophilia A mice injured with a needle puncture. FVIII−/− mice were administered with either EPCR-blocking (green circles) or EPCR-nonblocking mAb (red squares) (1 mg/kg). Twenty-four hours later, joint bleeding was induced with a needle puncture. Mice were monitored for 14 days to evaluate VBS (A) and changes in joint diameter (B). At the end of 14 days, mice were killed, and the limbs were photographed after removing overlying skin (C). UN, uninjured; I, injured. Data are mean ± SEM (n = 10 to 11 mice/group) (A-B). *P < .05; **P < 0.01; ***P < .001. (C) Pictures are representative images.
When joints were analyzed histopathologically 14 days after needle puncture, substantial synovial thickening with inflammation was observed in the injured knee (Figure 6A). The synovium was infiltrated with macrophages (Figure 6C) and remodeled with neovascularization (Figure 6E). Treatment of FVIII−/− mice with EPCR-blocking mAb markedly reduced the synovial hyperplasia (Figure 6A), macrophage invasion into the synovium (Figure 6C-D) and neovascularization (Figure 6E-F). Administration of EPCR-nonblocking mAb had no significant effect on the synovial hyperplasia, macrophage invasion, or neovascularization seen in joints following needle puncture–induced joint bleeding. Synovial iron deposition was detected in the injured knees of FVIII−/− mice. Treatment of mice prior to needle puncture with EPCR-blocking mAb prevented iron deposition in the synovium (supplemental Figure 7). Overall, immunohistopathological analysis of joint sections showed that the administration of EPCR-blocking mAb markedly reduced hemophilic synovitis (Figure 6B). Analysis of joint tissue sections for cartilage degeneration by Safranin O/Fast Green Staining revealed that administration of EPCR-blocking mAb prevented degeneration of cartilage induced by joint bleeding, whereas EPCR-nonblocking antibody treatment had no significant effect (Figure 7A-B). Staining joint tissues with Alcian blue, which also stains glycosaminoglycans and proteoglycans as with Safranin O/Fast Green, further supported the observation that EPCR-blocking antibody treatment reduces cartilage destruction induced by a joint bleed (supplemental Figure 8).

EPCR blocking reduces the severity of hemophilic synovitis following hemarthrosis in FVIII−/− mice. FVIII−/− mice were administered with EPCR-blocking mAb (bl. EPCR mAb), EPCR-nonblocking mAb (non-bl. EPCR mAb), or no mAb (No tr.). Twenty-four hours later, joint bleeding was induced with a needle puncture (I). UN indicates an uninjured knee. Two weeks after the injury, mice were killed, knee joints were excised, and joint tissue sections were processed for histopathological analysis by staining with H&E (A), macrophage marker F4/80 (C), and CD31 antibody to identify blood vessels (E). The red staining (C,E) indicates macrophages and blood vessels, respectively. (A) Two thin black arrows pointing each other indicate width of synovial lining layer, the yellow arrowhead points out red blood cells, and thick black arrows point out synovial villi. Arrows point out macrophages (C) and blood vessels (E). (B) Synovitis score. (D) Macrophage scoring. (F) Number of blood vessels counted per field. Shown are representative images, and data in bar graphs are mean ± SEM (n = 5 to 10 mice/group). ****P < .0001.
EPCR blocking reduces the severity of hemophilic synovitis following hemarthrosis in FVIII−/− mice. FVIII−/− mice were administered with EPCR-blocking mAb (bl. EPCR mAb), EPCR-nonblocking mAb (non-bl. EPCR mAb), or no mAb (No tr.). Twenty-four hours later, joint bleeding was induced with a needle puncture (I). UN indicates an uninjured knee. Two weeks after the injury, mice were killed, knee joints were excised, and joint tissue sections were processed for histopathological analysis by staining with H&E (A), macrophage marker F4/80 (C), and CD31 antibody to identify blood vessels (E). The red staining (C,E) indicates macrophages and blood vessels, respectively. (A) Two thin black arrows pointing each other indicate width of synovial lining layer, the yellow arrowhead points out red blood cells, and thick black arrows point out synovial villi. Arrows point out macrophages (C) and blood vessels (E). (B) Synovitis score. (D) Macrophage scoring. (F) Number of blood vessels counted per field. Shown are representative images, and data in bar graphs are mean ± SEM (n = 5 to 10 mice/group). ****P < .0001.

EPCR blocking prevents the degeneration of cartilage and elaboration of IL-6 in the synovium of FVIII−/− mice following hemarthrosis. FVIII−/− mice were administered with EPCR-blocking mAb (bl. EPCR mAb), EPCR-nonblocking mAb (non-bl. EPCR mAb), or no mAb (No tr.). Twenty-four hours later, joint bleeding was induced with a needle puncture (I). UN indicates an uninjured knee. (A) Two weeks after the injury, mice were killed, knee joints were excised, and joint tissue sections were stained with Safranin O/Fast Green Staining. Top panel images were captured at 4 magnification. The squared area was imaged at ×40 magnification (bottom panel). Arrows point out the articular cartilage. Loss of red stain in the articular cartilage region indicates the loss of glycosaminoglycans in the region and cartilage degeneration. (B) The quantified score of cartilage degeneration based on Safranin O/Fast Green Staining (0-3 scale; 0, normal/bright red staining of cartilage; 1, a slight reduction in staining; 2, a moderate reduction in staining; and 3, severe reduction/complete loss of staining). (C) One week after the injury, mice were killed, and synovial fluids were collected. IL-6 levels in synovial fluids were measured in enzyme-linked immunosorbent assay. The data shown are mean ± SEM (n = 4-7 mice/group). ***P < .001
EPCR blocking prevents the degeneration of cartilage and elaboration of IL-6 in the synovium of FVIII−/− mice following hemarthrosis. FVIII−/− mice were administered with EPCR-blocking mAb (bl. EPCR mAb), EPCR-nonblocking mAb (non-bl. EPCR mAb), or no mAb (No tr.). Twenty-four hours later, joint bleeding was induced with a needle puncture (I). UN indicates an uninjured knee. (A) Two weeks after the injury, mice were killed, knee joints were excised, and joint tissue sections were stained with Safranin O/Fast Green Staining. Top panel images were captured at 4 magnification. The squared area was imaged at ×40 magnification (bottom panel). Arrows point out the articular cartilage. Loss of red stain in the articular cartilage region indicates the loss of glycosaminoglycans in the region and cartilage degeneration. (B) The quantified score of cartilage degeneration based on Safranin O/Fast Green Staining (0-3 scale; 0, normal/bright red staining of cartilage; 1, a slight reduction in staining; 2, a moderate reduction in staining; and 3, severe reduction/complete loss of staining). (C) One week after the injury, mice were killed, and synovial fluids were collected. IL-6 levels in synovial fluids were measured in enzyme-linked immunosorbent assay. The data shown are mean ± SEM (n = 4-7 mice/group). ***P < .001
Blocking EPCR reduces IL-6 levels in the synovium following hemarthrosis
Hemarthrosis leads to a marked increase in IL-6 levels in the synovium (Figure 7C). Treatment of FVIII−/− mice with nonblocking EPCR mAb prior to induction of joint bleeding with needle puncture had no significant effect on the elaboration of IL-6 levels in the synovium. In contrast, the administration of EPCR-blocking mAb completely prevented the increase in IL-6 levels in the synovium following hemarthrosis (Figure 7C).
Discussion
The present study was prompted by our observations that pharmacological concentrations of rFVIIa, which is used successfully as a bypassing agent to treat hemophilia patients with inhibitors, also induce vascular barrier protective and anti-inflammatory effects.34-36 FVIIa-induced cytoprotective effects are dependent on its binding to EPCR and mediated via PAR1 activation.36,41 Since inflammation plays a key role in the pathogenesis of HA,4 we hypothesized that anti-inflammatory effects of FVIIa-EPCR would complement the hemostatic effect of rFVIIa in treating HA. Therefore, we expected that EPCR−/−FVIII−/− mice would be more prone to inflammation and synovitis following joint bleed and that rFVIIa treatment would be less effective in reducing HA in these mice compared with FVIII−/− and EPCR++ FVIII−/− mice. Surprisingly, we observed opposite results, ie, inflammation and synovitis were less severe in EPCR−/−FVIII−/− mice compared with FVIII−/− and EPCR++FVIII−/− mice and rFVIIa is more effective in preventing HA in EPCR−/−FVIII−/− mice compared with other groups.
The reduced severity of HA in EPCR−/−FVIII−/− is not due to reduced joint bleeding following needle puncture injury in these mice, as we found similar levels of blood in knee joints of all 3 groups of hemophilia mice, ie, FVIII−/−, EPCR++FVIII−/−, and EPCR−/−FVIII−/− mice. The reduced HA observed in EPCR−/−FVIII−/− mice, in which APC-mediated anticoagulant pathway is diminished, may reflect reduced recurrence of spontaneous joint bleeding in these mice following massive initial hemarthrosis induced by needle puncture. Earlier studies showed no substantial differences in thrombin-antithrombin (TAT) levels between WT and EPCR−/− mice at 2 months age, but significantly higher TAT levels were found in the EPCR−/− mice compared with WT mice when they were older (8 months old).42 We also did not find significant differences in basal TAT levels among FVIII−/−, EPCR++FVIII−/−, and EPCR−/−FVIII−/− mice (FVIII−/−, 7.1 ± 1.4; EPCR++FVIII−/−, 5.1 ± 1.0; and EPCR−/−FVIII−/−, 5.5 ± 0.21 ng/mL). Bleeding time/blood loss following the saphenous vein incision was also similar among 3 genotypes (data not shown). Nonetheless, it is likely that the absence of EPCR-dependent protein C activation in EPCR−/−FVIII−/− mice may lead to sufficient thrombin generation in the microvasculature to prevent bleeding into joints. Our earlier observation that showed the administration of neutralizing FVIII antibodies failed to induce severe acquired hemophilia phenotype in EPCR−/− mice as it did with WT mice33 supports the notion that low levels of thrombin are generated in EPCR-deficient mice in the absence (or only traces) of FVIII. Although the low levels of thrombin may not be sufficient to restore hemostasis in hemophilia following an acute injury such as the saphenous vein or tail cut, it may be sufficient in preventing recurrent joint bleeding in hemophilia mice. Our earlier studies showed that low levels of thrombin provide protection against vascular leakage in hemophilia A mice. In recent studies, Wyseure et al43 showed that joint bleeding was notably milder in acquired hemophilia A mice compared with FVIII−/− mice, and this could be due to defective thrombin-activatable fibrinolysis inhibitor (TAFI) activation in FVIII−/− mice. Their data also suggest that low levels of thrombin generated in acquired hemophilia A mice is sufficient to activate TAFI in the vascular bed of the joint, and this is at least in part responsible for milder phenotypic joint bleeding in acquired hemophilia compared with FVIII−/−. It is possible that thrombin generated in EPCR−/−FVIII−/− mice may be sufficient to activate TAFI in the vascular bed of the joint, and this may be responsible for milder HA observed in these mice compared with FVIII−/− mice.
Our recent studies showed that the extent of increase in inflammatory cytokines in response to LPS was slightly lower in EPCR-overexpressing mice compared with WT and EPCR-deficient mice, and FVIIa treatment markedly attenuated the LPS-induced cytokine elaboration in WT and EPCR-overexpressing mice, but not in EPCR-deficient mice.36 In contrast to these data, in the present study, we found that IL-6 levels in the synovium of injured knees of EPCR−/−FVIII−/− were markedly lower compared with FVIII−/− mice, whereas their levels were significantly higher in EPCR++FVIII−/− mice. These data should not be construed, as EPCR functions differently in the synovium than in other vascular beds. It is possible that the benefit of EPCR-mediated anti-inflammatory and barrier stabilization effects were negated by its anticoagulant function that blocks basal thrombin generation, which is critical in preventing spontaneous joint bleeding in severe FVIII deficiency.
Our present observation that rFVIIa is more effective in preventing HA in EPCR−/−FVIII−/− mice compared with FVIII−/− and EPCR++FVIII−/− mice was consistent with our earlier findings made in the saphenous vein injury model33 and supported the concept that EPCR levels influence the hemostatic effect of rFVIIa in treating hemophilia. These data further support our earlier hypothesis that rFVIIa displacement of protein C from EPCR that results in downregulation of APC generation and not the enhanced activation of FX by FVIIa bound to EPCR is the mechanism by which FVIIa interaction with EPCR contributes to the hemostatic effect of rFVIIa in hemophilia therapy.33,44
The current finding that blockade of EPCR function by a single administration of EPCR-blocking mAb is as effective as EPCR deficiency and better than rFVIIa treatment in reducing the HA in FVIII−/− mice identifies EPCR as a potential target in treating HA. In principle, the present data are consistent with recent observations that rebalancing hemostasis by reducing intrinsic anticoagulant activity is a viable option in treating hemophilia.45 Recent studies showed that a serpin that specifically inhibits APC-rescued hemostasis in a hemophilia mouse after tail-clip injury.46 It is reasonable to conclude that the therapeutic effect of EPCR-blocking mAb comes from the downregulation of APC generation that rebalances hemostasis (supplemental Figure 9). However, we cannot completely exclude other possibilities. EPCR, in addition to binding protein C/APC and FVII/FVIIa, also binds other ligands, including the ligands that associate with macrophages and T cells.44 Furthermore, EPCR has been identified as a stem cell marker and plays a role in the repopulation and homing of hematopoietic stem cells.47,48 Acharya et al18 showed the presence of bone marrow–derived endothelial progenitor cells in the synovium of hemophilic joint disease patients and suggested that these cells may play a role in neoangiogenesis associated with HA. Therefore, it is conceivable that, in addition to rebalancing hemostasis, EPCR-blocking mAb could block other EPCR-mediated cellular processes that contribute to HA. In this context, it is pertinent to point out that the administration of EPCR-blocking mAb, although shortened the bleeding time in moderate hemophilia A induced by the antibody,33 it failed to correct bleeding in FVIII−/− mice subjected to the saphenous vein injury.49 The differences in the effectiveness of EPCR mAb in preventing saphenous vein injury–induced bleeding and joint bleeding in FVIII−/− mice may reflect differences in threshold levels of thrombin necessary to exert hemostatic effect against saphenous vein bleeding vs joint bleeding or differences in the generation of APC in different vascular beds.
Since EPCR was shown to support APC- and FVIIa-induced cytoprotective signaling,44,50 it may appear counterintuitive to suggest that inhibition of EPCR could be a viable treatment option in preventing joint disease in hemophilia patients. Here, it is important to note that there is no evidence that EPCR is essential for health except that extraembryonic expression of EPCR may be necessary for embryonic viability.42 EPCR-deficient mice were found to be healthy, and no hemorrhage or visible evidence of spontaneous thrombosis was observed.42 In our observations of EPCR-deficient mice over the past 10 years, we have found no evidence of health-related problems. It may be pertinent to note here that almost all the studies that demonstrated the beneficial effects of EPCR were from disease-induced model systems where APC or FVIIa were administered.41,44,50 In our earlier studies, in the absence of FVIIa treatment, we found no significant differences in the extent of inflammation between EPCR-deficient and WT mice in response to LPS challenge.35,36 Therefore, it is unlikely that the administration of EPCR mAb to prevent HA will have any negative consequences on the health of hemophilia patients. The prolonged protective effect of the EPCR antibody, its robust effectiveness in reducing joint bleeding and hemophilic synovitis, and the possibility that it could be used to treat all categories of hemophilia make it an attractive option.
The online version of this article contains a data supplement.
The publication costs of this article were defrayed in part by page charge payment. Therefore, and solely to indicate this fact, this article is hereby marked “advertisement” in accordance with 18 USC section 1734.
Acknowledgments
The authors thank Jagan Sundaram and Anuradha Rajulapati, former staff in the laboratory, for their contribution to the generation of EPCR++FVIII−/− and EPCR−/−FVIII−/− mice. They also acknowledge the contribution of Daniel Hernandez for scoring visual bleeding and Sinchana Basoor for scoring immunohistochemistry images. The authors appreciate the help of Shiva Keshava for his guidance on some of the experimental procedures and reviewing images.
This work was supported by National Institutes of Health/National Heart, Lung, and Blood Institute grants HL107483 (L.V.M.R.) and UM1 HL120877 (C.T.E.).
Authorship
Contribution: J.M. performed all experiments and summarized data; U.R.P. contributed to the study design, data review, and analysis; C.T.E. provided breeding pairs of EPCR-deficient and Tie2-EPCR mice and mouse EPCR–specific blocking and nonblocking monoclonal antibodies; L.V.M.R. conceived and designed the research, analyzed the data, and wrote the manuscript; and all authors contributed to the preparation of the final version of the manuscript.
Conflict-of-interest disclosure: The authors declare no competing financial interests.
Correspondence: L. Vijaya Mohan Rao, Department of Cellular and Molecular Biology, The University of Texas Health Science Center at Tyler, 11937 US Highway 271, Tyler, TX 75708-3154; e-mail: vijay.rao@uthct.edu.

This feature is available to Subscribers Only
Sign In or Create an Account Close Modal