

Key Points
Transforming MPL exon 10 mutations are not limited to S505N and W515L/K/R/A, but are highly restricted by position and amino acid type.
Many second-site mutations can enhance constitutive signaling by a canonical MPL exon 10 mutation and several are found in MPN patients.

Abstract
The single transmembrane domain (TMD) of the human thrombopoietin receptor (TpoR/myeloproliferative leukemia [MPL] protein), encoded by exon 10 of the MPL gene, is a hotspot for somatic mutations associated with myeloproliferative neoplasms (MPNs). Approximately 6% and 14% of JAK2 V617F− essential thrombocythemia and primary myelofibrosis patients, respectively, have “canonical” MPL exon 10 driver mutations W515L/K/R/A or S505N, which generate constitutively active receptors and consequent loss of Tpo dependence. Other “noncanonical” MPL exon 10 mutations have also been identified in patients, both alone and in combination with canonical mutations, but, in almost all cases, their functional consequences and relevance to disease are unknown. Here, we used a deep mutational scanning approach to evaluate all possible single amino acid substitutions in the human TpoR TMD for their ability to confer cytokine-independent growth in Ba/F3 cells. We identified all currently recognized driver mutations and 7 novel mutations that cause constitutive TpoR activation, and a much larger number of second-site mutations that enhance S505N-driven activation. We found examples of both of these categories in published and previously unpublished MPL exon 10 sequencing data from MPN patients, demonstrating that some, if not all, of the new mutations reported here represent likely drivers or modifiers of myeloproliferative disease.
Introduction
The thrombopoietin (Tpo) receptor (TpoR/myeloproliferative leukemia [MPL] protein) regulates hematopoietic stem cell maintenance, megakaryocyte development, and platelet production.1 Its transmembrane domain (TMD), encoded by exon 10 of the MPL gene, is a hotspot for mutations associated with myeloproliferative neoplasms (MPNs) essential thrombocythemia (ET) and primary myelofibrosis.2 Approximately 6% and 14% of JAK2 V617F− ET/primary myelofibrosis patients have “canonical” MPL exon 10 mutations3 W515L/K/R/A4,5 or S505N6-8 that cause gain-of-function defects leading to constitutive activation of JAK/STAT signaling and platelet overproduction. W515L/K/R/A mutations destroy an inhibitory site that prevents activation when Tpo is not bound,5,9-11 whereas S505N activates TpoR by driving strong dimerization.6,12,13 Other “noncanonical” MPL exon 10 mutations have been identified in patients,14-20 but, in most cases, their functional consequences and relevance to disease are unknown. Here, we report 2 deep-sequencing–based saturation mutagenesis screens in which we examined all possible single amino acid substitutions in the TpoR TMD and identified novel mutations that cause constitutive TpoR activation or enhance S505N-driven activation in Ba/F3 cells.
Study design
Deep mutational scanning (DMS)21 libraries, prepared as described (supplemental Methods; supplemental Figures 1 and 2 [available on the Blood Web site]), were retrovirally expressed in interleukin 3 (IL-3)-dependent Ba/F3 cells and selected for factor-free growth by 48 hours of culture without IL-3 or any other cytokines. TpoR complementary DNA prepared from selected and unselected populations was Illumina sequenced to identify and quantitate variants. Changes in variant frequencies are summarized as Enrich222 scores and presented in sequence-function heat maps where red indicates enrichment and blue indicates depletion of each receptor variant after culture without IL-3. Details of sequencing data analysis for the screens and growth/signaling assays for individual variants can be found in figure legends and supplemental Methods. MPL exon 10 sequencing data from 2452 MPN patients was reviewed to identify mutations that showed transforming or enhancing effects in our screens.
Results and discussion
To identify novel drivers and modifiers of MPNs, we performed 2 DMS screens designed to identify all single amino acid substitutions in the TpoR TMD that conferred factor-free growth or enhanced the growth phenotype of S505N in Ba/F3 cells. In the first screen, 24 of the 580 TpoR variants examined were enriched relative to wild type (WT) upon cytokine withdrawal (Figure 1A red squares), indicating constitutive receptor activation. All other variants were depleted (Figure 1A white to blue squares) and therefore did not result in constitutive activity. Enriched variants included the expected S505N6,7 and many different substitutions at W515.4,5,9 In addition, S493L/C, L498W, V501S/A, L502S/C, and L508Q were significantly enriched. H499C and V501G showed possible weak enrichment. Each of these variants was isolated from the DMS library or regenerated using traditional site-directed mutagenesis, resequenced, and tested separately for growth in IL-3, Tpo, or no cytokine (Figure 1B). Cells expressing strongly enriched variants were responsive to Tpo (Figure 1B green bars) and grew over 2 days with no cytokines (Figure 1B red bars), whereas WT TpoR, H499C, and V501G cells died upon cytokine withdrawal. Several of the newly identified variants (L498W, V501S, V501A, and L502S) showed significant STAT5 and/or extracellular signal-regulated kinase 1/2 (ERK1/2) phosphorylation in the absence of cytokines (Figure 1C and D; supplemental Figure 3). Enrichment scores from the DMS screen and cell growth in the independent assay were strongly correlated (Figure 1E), confirming that variant counts in the mixed cell population from our DMS screen accurately reflected the strength of individual variant phenotypes.
![Transforming TpoR TMD mutations are few in number and are highly restricted by position and substituting amino acid type. (A) Sequence-function map showing the results of saturation mutagenesis of human TpoR TMD (residues 488-516) in Ba/F3 cells. WT sequence and amino acid number in the mature polypeptide are shown to the right and left of the heat map, respectively. Each square reflects the combined data from all synonymous variants at that position. Color of each square indicates the log ratio enrichment score for variant frequencies in Ba/F3 cells cultured for 48 hours without cytokines compared with the unselected library, according to the scale below the map. Scores are relative to WT (white squares with dots; score of 0). Red shades indicate enrichment. Blue shades indicate depletion. Diagonal lines are proportional to standard deviations of scores across 6 replicate selections. TMD helix model shows the predicted locations of sites where mutations caused constitutive activity, colored by approximate enrichment score. (B) Selected variants from (A) were transduced individually into Ba/F3 cells and tested for growth in IL-3, Tpo (100 ng/ml), or no cytokine (RPMI-1640). Data are plotted as fold-change over starting cell density (dotted line) after 48 hours. Individual data points represent the mean of technical triplicates from each of 3 independent experiments. Error bars indicate standard deviation. No statistically significant differences were observed in IL-3 or Tpo; significance values are indicated for all variants compared with WT in no cytokine (1-way analysis of variance [ANOVA]: ****P < .0001). (C) Western blot analysis of STAT5 and ERK1/2 activation status. Ba/F3 cells expressing each variant were serum/cytokine starved for 7 hours prior to treatment with 50 ng/mL recombinant human Tpo (hTpo; WT only) or without cytokines for 30 minutes. Lysates were separated by sodium dodecyl sulfate–polyacrylamide gel electrophoresis (SDS-PAGE) and immunoblotted with antibodies against the indicated targets. *Nonspecific background band in phosphorylated ERK 1/2 (pERK1/2) immunoblots; #glycosylation variants of hemagglutinin (HA)-tagged human TpoR (hTpoR) protein. (D) Densitometric analysis of phosphorylated STAT5 (pSTAT5) and pERK1/2 signal from 3 independent experiments performed as shown in (C). All immunoblots are shown in supplemental Figure 2. Signal intensities are reported as fold-increase over WT with no cytokine treatment (indicated by dotted line at 1). Significance values are indicated for all variants compared with the inactive V501G variant for pSTAT5 or pERK1/2 (1-way ANOVA: ****P < .0001; ***P < .001; *P < .05). (E) Plot shows correlation between log ratio enrichment scores in the DMS screen (A) and cell counts from independent growth experiments (B). Error bars indicate the standard deviations of enrichment scores calculated in Enrich2 (y-axis) and standard deviation of final cell counts in the experiment from panel (B) (x-axis). ns, not significant.](https://ash.silverchair-cdn.com/ash/content_public/journal/blood/135/4/10.1182_blood.2019002561/5/m_bloodbld2019002561f1.png?Expires=1769084576&Signature=lOFw~EoKlWB5O182IV~dZm2IfeilkN1MoGymy8r9q0wGniyFRHAc0GuhI7cI6YIL5AFJSSbaZAuHFaIxIBREddtVyVOeQF75ivfxfJa4q5CyG4xQ2lpeKthfdDItYQAvcVdoHL8i0TxwiWQDHmtiYRTFqYFSvgKUfYVtAe9aFM6Z8Y3gY0xi9Kf071jPk4~yJk3gplcb538F3NSOm1iaDtc1CaLoAnyjiN~GY-XBY67EdOSNYgZ-41Evp9dYaYRyjpZ9b0T2z1ZcDVS6CXHn-Q~K25TeuaLkY8AOJhZ6nKlnyNLbqhLAJTyI-LmMAE2Ko9v~5et9EBA9Ss7cK12TKg__&Key-Pair-Id=APKAIE5G5CRDK6RD3PGA)
Transforming TpoR TMD mutations are few in number and are highly restricted by position and substituting amino acid type. (A) Sequence-function map showing the results of saturation mutagenesis of human TpoR TMD (residues 488-516) in Ba/F3 cells. WT sequence and amino acid number in the mature polypeptide are shown to the right and left of the heat map, respectively. Each square reflects the combined data from all synonymous variants at that position. Color of each square indicates the log ratio enrichment score for variant frequencies in Ba/F3 cells cultured for 48 hours without cytokines compared with the unselected library, according to the scale below the map. Scores are relative to WT (white squares with dots; score of 0). Red shades indicate enrichment. Blue shades indicate depletion. Diagonal lines are proportional to standard deviations of scores across 6 replicate selections. TMD helix model shows the predicted locations of sites where mutations caused constitutive activity, colored by approximate enrichment score. (B) Selected variants from (A) were transduced individually into Ba/F3 cells and tested for growth in IL-3, Tpo (100 ng/ml), or no cytokine (RPMI-1640). Data are plotted as fold-change over starting cell density (dotted line) after 48 hours. Individual data points represent the mean of technical triplicates from each of 3 independent experiments. Error bars indicate standard deviation. No statistically significant differences were observed in IL-3 or Tpo; significance values are indicated for all variants compared with WT in no cytokine (1-way analysis of variance [ANOVA]: ****P < .0001). (C) Western blot analysis of STAT5 and ERK1/2 activation status. Ba/F3 cells expressing each variant were serum/cytokine starved for 7 hours prior to treatment with 50 ng/mL recombinant human Tpo (hTpo; WT only) or without cytokines for 30 minutes. Lysates were separated by sodium dodecyl sulfate–polyacrylamide gel electrophoresis (SDS-PAGE) and immunoblotted with antibodies against the indicated targets. *Nonspecific background band in phosphorylated ERK 1/2 (pERK1/2) immunoblots; #glycosylation variants of hemagglutinin (HA)-tagged human TpoR (hTpoR) protein. (D) Densitometric analysis of phosphorylated STAT5 (pSTAT5) and pERK1/2 signal from 3 independent experiments performed as shown in (C). All immunoblots are shown in supplemental Figure 2. Signal intensities are reported as fold-increase over WT with no cytokine treatment (indicated by dotted line at 1). Significance values are indicated for all variants compared with the inactive V501G variant for pSTAT5 or pERK1/2 (1-way ANOVA: ****P < .0001; ***P < .001; *P < .05). (E) Plot shows correlation between log ratio enrichment scores in the DMS screen (A) and cell counts from independent growth experiments (B). Error bars indicate the standard deviations of enrichment scores calculated in Enrich2 (y-axis) and standard deviation of final cell counts in the experiment from panel (B) (x-axis). ns, not significant.
Transforming TpoR TMD mutations are few in number and are highly restricted by position and substituting amino acid type. (A) Sequence-function map showing the results of saturation mutagenesis of human TpoR TMD (residues 488-516) in Ba/F3 cells. WT sequence and amino acid number in the mature polypeptide are shown to the right and left of the heat map, respectively. Each square reflects the combined data from all synonymous variants at that position. Color of each square indicates the log ratio enrichment score for variant frequencies in Ba/F3 cells cultured for 48 hours without cytokines compared with the unselected library, according to the scale below the map. Scores are relative to WT (white squares with dots; score of 0). Red shades indicate enrichment. Blue shades indicate depletion. Diagonal lines are proportional to standard deviations of scores across 6 replicate selections. TMD helix model shows the predicted locations of sites where mutations caused constitutive activity, colored by approximate enrichment score. (B) Selected variants from (A) were transduced individually into Ba/F3 cells and tested for growth in IL-3, Tpo (100 ng/ml), or no cytokine (RPMI-1640). Data are plotted as fold-change over starting cell density (dotted line) after 48 hours. Individual data points represent the mean of technical triplicates from each of 3 independent experiments. Error bars indicate standard deviation. No statistically significant differences were observed in IL-3 or Tpo; significance values are indicated for all variants compared with WT in no cytokine (1-way analysis of variance [ANOVA]: ****P < .0001). (C) Western blot analysis of STAT5 and ERK1/2 activation status. Ba/F3 cells expressing each variant were serum/cytokine starved for 7 hours prior to treatment with 50 ng/mL recombinant human Tpo (hTpo; WT only) or without cytokines for 30 minutes. Lysates were separated by sodium dodecyl sulfate–polyacrylamide gel electrophoresis (SDS-PAGE) and immunoblotted with antibodies against the indicated targets. *Nonspecific background band in phosphorylated ERK 1/2 (pERK1/2) immunoblots; #glycosylation variants of hemagglutinin (HA)-tagged human TpoR (hTpoR) protein. (D) Densitometric analysis of phosphorylated STAT5 (pSTAT5) and pERK1/2 signal from 3 independent experiments performed as shown in (C). All immunoblots are shown in supplemental Figure 2. Signal intensities are reported as fold-increase over WT with no cytokine treatment (indicated by dotted line at 1). Significance values are indicated for all variants compared with the inactive V501G variant for pSTAT5 or pERK1/2 (1-way ANOVA: ****P < .0001; ***P < .001; *P < .05). (E) Plot shows correlation between log ratio enrichment scores in the DMS screen (A) and cell counts from independent growth experiments (B). Error bars indicate the standard deviations of enrichment scores calculated in Enrich2 (y-axis) and standard deviation of final cell counts in the experiment from panel (B) (x-axis). ns, not significant.
Like S505N, the newly identified constitutively active substitutions exhibited stringent amino acid specificity, with only 1 or 2 substitutions enriched at any position. Notably, V501A has been documented in MPN patients15,16,18,20 and was recently shown to drive constitutive TpoR activation, whereas V501G did not.23 Our data show that this relationship holds in the context of cytokine-independent cell growth and that V501S confers a significantly stronger phenotype. With the exceptions of the extracellular juxtamembrane S493L/C and intracellular juxtamembrane W515 variants, all constitutively activating substitutions were located on the same face of an α-helix (Figure 1A), suggesting that they may support formation of structurally similar TpoR TMD dimer interfaces.11,13
Previous studies have identified patients with a canonical MPL exon 10 mutation and additional exon 10 mutations on the same allele.18,19 Whether these confer qualitatively or quantitatively different cellular phenotypes from the single canonical mutations is unclear. To test how second-site mutations in the TpoR TMD may modulate the activity of a known driver mutation, we performed a second DMS screen on the S505N background (Figure 2A), often found as a germline mutation in hereditary thrombocythemia.6,8,24 As expected, substitution of any residue other than asparagine at position 505 resulted in loss of constitutive activity. However, >90 different second-site mutations were enriched compared with S505N alone. These included multiple W515 substitutions, indicating that elimination of this inhibitory residue is enhancing as well as independently transforming. R514K and many substitutions at Q516 were also enriched, showing that even changes close to W515 can have significant effects as second-site mutations.
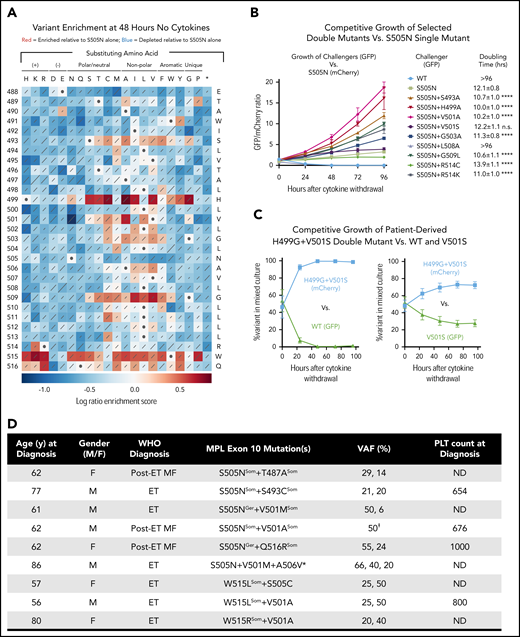
A broad range of second-site TpoR TMD mutations enhance the cytokine-independent activity of the S505N mutant receptor. (A) Sequence-function map showing the results of saturation mutagenesis of human TpoR-S505N TMD (residues 488-516). Color of each square indicates the log ratio enrichment score for variant frequencies in Ba/F3 cells cultured for 48 hours without cytokines compared with the unselected library, according to the scale below the map. Scores reflect variant frequency changes relative to S505N (white squares with dots; score of 0). Red shades indicate enrichment. Blue shades indicate depletion. Diagonal lines are proportional to standard deviations of scores across 6 replicate selections. (B) A selection of double mutants from (A) were tested in a competition assay in which TpoR-S505N (reference) cells expressed mCherry and double mutants (challengers) expressed green fluorescent protein (GFP). Approximately equal numbers of cells (GFP-to-mCherry ratio ∼1) were coseeded in the same well and grown for 96 hours without cytokines. Relative levels of GFP+ and mCherry+ cells were determined daily by flow cytometry and are plotted as the GFP-to-mCherry ratio. Data points represent the mean of 3 technical replicates from 1 of 3 independent experiments performed. Error bars show standard deviations. Data for additional experiments are shown in supplemental Figure 3. Doubling times (mean and standard deviation) were calculated using combined data from all 3 independent experiments. Significance values are given for each challenger’s doubling time compared with that of S505N (1-way ANOVA: ****P < .0001). Doubling times >96 hours indicate rapid death or no measurable growth. (C) Competition assay performed as in panel B testing the patient-derived TpoR H499G+V501S27 double mutant (mCherry) coseeded with GFP-expressing WT (left) or V501S (right). Plots show percentage of GFP and mCherry cells in the mixed cultures over 96 hours of growth. Data points represent the mean of 3 independent experiments. Error bars are standard deviations. (D) MPN patients identified with multiple MPL exon 10 mutations. (E) One variant allele frequency (VAF) value for this patient reflects the frequency of the paired mutations. *Patient with triple MPL exon 10 mutation was also JAK2-V617F+ (VAF 100%). All other patients were WT at JAK2 and CALR exon 9. Where VAF values, knowledge of familial disease, and/or availability of lymphocyte DNA sequences allow differentiation between germline (Ger) and somatic (Som) lesions, individual mutations are marked in superscript. F, female; M, male; MF, myelofibrosis; ND, not determined; PLT, platelet count at diagnosis (×106/mL).
A broad range of second-site TpoR TMD mutations enhance the cytokine-independent activity of the S505N mutant receptor. (A) Sequence-function map showing the results of saturation mutagenesis of human TpoR-S505N TMD (residues 488-516). Color of each square indicates the log ratio enrichment score for variant frequencies in Ba/F3 cells cultured for 48 hours without cytokines compared with the unselected library, according to the scale below the map. Scores reflect variant frequency changes relative to S505N (white squares with dots; score of 0). Red shades indicate enrichment. Blue shades indicate depletion. Diagonal lines are proportional to standard deviations of scores across 6 replicate selections. (B) A selection of double mutants from (A) were tested in a competition assay in which TpoR-S505N (reference) cells expressed mCherry and double mutants (challengers) expressed green fluorescent protein (GFP). Approximately equal numbers of cells (GFP-to-mCherry ratio ∼1) were coseeded in the same well and grown for 96 hours without cytokines. Relative levels of GFP+ and mCherry+ cells were determined daily by flow cytometry and are plotted as the GFP-to-mCherry ratio. Data points represent the mean of 3 technical replicates from 1 of 3 independent experiments performed. Error bars show standard deviations. Data for additional experiments are shown in supplemental Figure 3. Doubling times (mean and standard deviation) were calculated using combined data from all 3 independent experiments. Significance values are given for each challenger’s doubling time compared with that of S505N (1-way ANOVA: ****P < .0001). Doubling times >96 hours indicate rapid death or no measurable growth. (C) Competition assay performed as in panel B testing the patient-derived TpoR H499G+V501S27 double mutant (mCherry) coseeded with GFP-expressing WT (left) or V501S (right). Plots show percentage of GFP and mCherry cells in the mixed cultures over 96 hours of growth. Data points represent the mean of 3 independent experiments. Error bars are standard deviations. (D) MPN patients identified with multiple MPL exon 10 mutations. (E) One variant allele frequency (VAF) value for this patient reflects the frequency of the paired mutations. *Patient with triple MPL exon 10 mutation was also JAK2-V617F+ (VAF 100%). All other patients were WT at JAK2 and CALR exon 9. Where VAF values, knowledge of familial disease, and/or availability of lymphocyte DNA sequences allow differentiation between germline (Ger) and somatic (Som) lesions, individual mutations are marked in superscript. F, female; M, male; MF, myelofibrosis; ND, not determined; PLT, platelet count at diagnosis (×106/mL).
Most other enhancing substitutions were concentrated at sites that showed little (S493) or no constitutive activity (H499, G503, G509) in the first screen and the substituting amino acids were generally more hydrophobic than the native ones. H499 substitutions were particularly strong enhancers, consistent with the reported role of this residue in suppressing spontaneous TpoR activation.25,26 Interestingly, the pattern of enhancing substitutions at V501 reveals a different amino acid hierarchy from the first screen: although V501S provided the strongest constitutive activity as a single substitution, V501A is a much stronger enhancer on the S505N background. A selection of double mutants tested in direct cell growth competition assays (Figure 2B; supplemental Figure 4) showed that enriched variants reliably outgrew S505N in mixed cultures, whereas WT and depleted variants were rapidly overtaken. Thus, in contrast to the small number of constitutively active variants in the single-site screen, our second-site screen reveals that a much larger number of substitutions can exacerbate the effects of an independently transforming mutation. These effects may arise from several different mechanisms, and further studies will be required to define their modes of action. We also note that many second-site substitutions appear to antagonize S505N-driven activation (Figure 2B blue squares), though our screen does not distinguish between reduced activity and reduced expression.
A survey of MPL exon 10 sequencing data from MPN patients shows that the results of our screens are highly relevant for identifying clinically significant mutations. Of 9 noncanonical TpoR TMD mutations reported in the COSMIC database (cancer.sanger.ac.uk), 1 (V501A) caused cytokine-independent growth in our screens and an additional 5 (L500V, V501L, V507I, G509C, and R514K) significantly enhanced the cytokine-independent growth of S505N. A recently published case report27 describes an ET patient with a 9-nucleotide germline insertion/deletion resulting in a H499G+V501S double mutation. V501S is strongly activating (Figure 1) and H499G is among the strongest enhancers on the S505N background (Figure 2A). A competition experiment shows it has a similar enhancing effect on V501S (Figure 2B), and the H499G+V501S double mutant even confers a stronger growth phenotype than S505N alone (supplemental Figure 4). Finally, in a survey of unpublished data from our ongoing MPN patient sequencing efforts (Figure 2D), we have identified patients with S505N+T487A, S505N+S493C, S505N+V501M, S505N+V501A, and S505N+Q516R double mutations as well as 1 patient with a S505N+V501M+A506V triple mutation. Although position 487 was not randomized in our screen, T487A is a known activating mutation associated with acute megakaryoblastic leukemia.28 The remaining patient-derived second-site mutations were all identified as enhancers of S505N in our second-site screen and are therefore likely to be particularly strong MPN drivers. Additional published reports of patients with double mutations include V501A+W515R,18 V501A+W515L,18 and S505C+W515L,18,19 and we identified a second instance of each of these combinations in our patient cohorts (Figure 2D). Finally, we note that any of these mutations could occur on the background of additional mutations in other domains of TpoR that are known to modulate function, such as P106L or S204P/F in the extracellular domain and Y591F/N in the intracellular cytoplasmic tail,2 which we have not evaluated here. In light of our results, we conclude that all noncanonical MPL exon 10 mutations identified in patients should be closely examined as potential drivers or modifiers of oncogenic transformation. The data reported here provide a valuable diagnostic reference to evaluate their potential functional implications.
The raw reads reported in this article have been deposited in the Sequence Read Archive (SRA; www.ncbi.nlm.nih.gov/sra) database (accession number SRR10193506). The processed scores and counts reported in this article have been deposited in the Multiplexed Assays of Variant Effect Database (MaveDB; www.mavedb.org) (accession number urn:mavedb:00000043).
The online version of this article contains a data supplement.
The publication costs of this article were defrayed in part by page charge payment. Therefore, and solely to indicate this fact, this article is hereby marked “advertisement” in accordance with 18 USC section 1734.
Acknowledgments
The authors thank Warren Alexander, Andrew Roberts, and Douglas Fowler for helpful discussions.
This work was supported by the Australian National Health and Medical Research Council (NHMRC; project grant 115734, program grant 1054618, and Independent Research Institute Infrastructure Support Scheme [IRIISS] infrastructure support) and Victorian State Government Operational Infrastructure Support (to Walter and Eliza Hall Institute [WEHI]). Research was carried out in part at the Translational Research Institute (Woolloongabba, QLD, Australia), which is supported by a grant from the Australian government. Molecular characterization of patients followed at the University of Florence and the University of Pavia in Italy was supported by Associazione Italiana per la Ricerca sul Cancro (AIRC; 5x1000) call “Metastatic disease: the key unmet need in oncology” to the MYNERVA project (#21267).
Authorship
Contribution: M.J.C. and M.E.C. conceived and jointly supervised the study; A.F.R., S.W., and M.J.C. designed the library production and DMS sequencing strategies; J.L.B., S.M.L., C.M.M.L., and Y.C. performed and analyzed screening and validation experiments; A.J.B. constructed the original expression vectors and analyzed signaling data; S.W. performed Illumina sequencing; J.L.B., S.M.L., and M.J.C. processed and analyzed DMS sequencing data; P.G., E.R., D.P., M.C., and A.M.V. collected patient samples and analyzed clinical sequencing data; M.E.C., M.J.C., and J.L.B. wrote the paper; and all authors reviewed and edited drafts of the manuscript.
Conflict-of-interest disclosure: The authors declare no competing financial interests.
Correspondence: Melissa J. Call, Walter and Eliza Hall Institute of Medical Research, 1G Royal Parade, Parkville, VIC 3052 Australia; e-mail: mjcall@wehi.edu.au; or Matthew E. Call, Walter and Eliza Hall Institute of Medical Research, 1G Royal Parade, Parkville, VIC 3052 Australia; e-mail: mecall@wehi.edu.au.

This feature is available to Subscribers Only
Sign In or Create an Account Close Modal