

In this issue of Blood, demonstrate that preventing cyclophilin D (CypD)-mediated platelet necrosis protects neurons from death in a model of ischemic stroke with reperfusion.1 This decrease in neuronal cell death coincides with decreased formation of CypD-dependent necrotic platelet/neutrophil aggregates.
Mice without CypD in their platelets are protected from reperfusion injury following cerebral ischemia. See Figure 1A-B in the article by Denorme et al that begins on page 429.
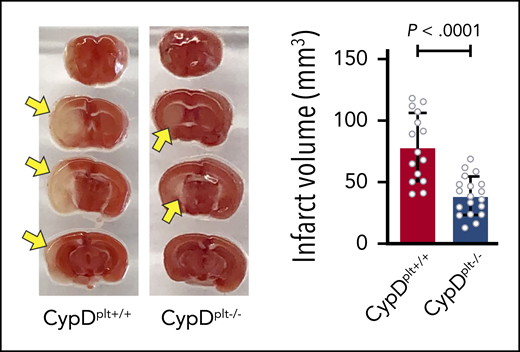
Mice without CypD in their platelets are protected from reperfusion injury following cerebral ischemia. See Figure 1A-B in the article by Denorme et al that begins on page 429.
CypD is a mitochondrial peptidylprolylisomerase. Its inhibition or deletion abrogates the formation of a large inner mitochondrial membrane pore known as the mitochondrial permeability transition pore (mPTP). Prolonged mPTP formation results in necrotic cell death, and pharmacologic agents that inhibit CypD and mPTP formation remain an interesting target for the prevention of cell death following ischemic injury.2 In platelets, CypD and mPTP formation mediate the transition of an activated platelet from a classical aggregatory phenotype to a procoagulant phenotype.3 These procoagulant, or necrotic, platelets have distinct phenotypic and functional characteristics. Among these is the procoagulant platelet’s affinity for neutrophils, and their role in the initiation and formation of platelet/neutrophil aggregates.4
Rapid vessel recanalization remains the mainstay of therapy in arterial thrombosis. However, following successful reperfusion, injured cells within the area of previously reduced blood flow are at risk of death. Within this at-risk area, ischemic damage and reactive oxygen species have been proposed as key contributors to cell death. Platelets and neutrophils have similarly been implicated, but the pathologic mechanism by which they mediate cell death in reperfused tissue remains controversial.5 Interestingly, blockade of neither the major platelet receptor integrin αIIbβ3 nor the neutrophil endothelial receptor ICAM-1 prevents reperfusion injury.6
Using PF4-Cre mice with a floxed CypD allele, Denorme et al tested the role of platelet necrosis in murine stroke. Mice without platelet CypD were protected from reperfusion injury measured 24 hours after resumption of flow. Specifically, in platelet CypD null mice, the area of ischemic injury was substantially reduced, and brain function was spared; this coincided with improved recovery of blood flow to the treated hemisphere (see figure). In mice with permanent ischemia (no reperfusion), the protective effect of ablating platelet CypD was not observed demonstrating that the protection offered by platelet CypD deletion functions only in the context of reperfusion. Aggregates of necrotic platelets (phosphatidylserine positive) and neutrophils (necPlt/Neut Aggs) were increased in mice with ischemic brain injury in the circulation and within the injured brain hemisphere. This increase in necPlt/Neut Aggs was decreased in platelet CypD null mice. To test the pathologic role of necPlt/Neut Aggs, mice with and without platelet CypD were depleted of neutrophils and subjected to ischemia/reperfusion injury. Neutrophil depletion resulted in decreased neuronal damage, and this was associated with improved recovery of blood flow. However, in the context of neutrophil depletion, no additional protection was provided by the absence of platelet CypD, strongly implying that the protective effects of neutrophil and platelet CypD depletion are mediated by prevention of necPlt/Neut Agg formation.
Recently, the formation of CypD-mediated necPlt/Neut Aggs was shown to mediate distal vasoocclusive tissue injury within the lung following reperfusion of the ischemic gut.7 Together with the results presented by Denorme et al, these studies place the necPlt/Neut Agg at the center of the pathologic manifestations of ischemia/reperfusion injury and point toward alternative therapeutic approaches for the treatment of reperfusion injury. The initiating mediator of necrotic platelet formation following reperfusion is yet to be determined. In vitro, thrombin and collagen initiate procoagulant, or necrotic, platelet formation in a fraction of activated platelets. The reactive oxygen species H2O2 can increase procoagulant platelet formation in thrombin-stimulated platelets in vitro,8 and reactive oxygen species are increased in reperfused tissues. Increased necPlt/Neut Aggs are associated with decreased blood flow in reperfused tissues, but it remains to be established whether this is due to a direct occlusive effect of lodged aggregates or a secondary vasoconstrictive or cell-damaging effect as might be initiated by the production of novel paracrine mediators. Finally, it will be interesting to investigate whether CypD-mediated necPlt/Neut Agg formation plays a similar central role in reperfusion injury in other tissues, including the myocardium.
These studies should reinvigorate interest in both the mPTP and the necrotic platelet as a therapeutic target to limit ischemia reperfusion injury. Mice without CypD or platelet CypD do not have a primary hemostatic defect, suggesting that limiting necrotic platelet formation may provide a novel nonhemorrhagic platelet-targeted therapy. Inhibition of mPTP formation prevents necrotic platelet formation, but has no effect on platelet aggregation or granule release.3 In stroke patients, cyclosporine treatment resulted in a substantial increase in neuronal recovery in a subgroup analysis of patients with successful thrombolytic reperfusion.9 In patients with transient neuronal ischemia, procoagulant platelet potential, a measure of the fraction of platelets that become procoagulant following standardized agonist stimulation is directly correlated with risk of stroke in the next 30 days.10 Based on this result, it may be possible to risk-stratify therapy toward those patients with higher risk of downstream, ischemic injury. Future studies focusing on more specific inhibitors of CypD or alternative targets related to necrotic platelet formation may prove to be more effective in limiting reperfusion injury.
Conflict-of-interest disclosure: The author declares no competing financial interests.

This feature is available to Subscribers Only
Sign In or Create an Account Close Modal