


Abstract
Viral infections are common and are potentially life-threatening in patients with moderate to severe primary immunodeficiency disorders. Because T-cell immunity contributes to the control of many viral pathogens, adoptive immunotherapy with virus-specific T cells (VSTs) has been a logical and effective way of combating severe viral disease in immunocompromised patients in multiple phase 1 and 2 clinical trials. Common viral targets include cytomegalovirus, Epstein-Barr virus, and adenovirus, though recent published studies have successfully targeted additional pathogens, including HHV6, BK virus, and JC virus. Though most studies have used VSTs derived from allogenic stem cell donors, the use of banked VSTs derived from partially HLA-matched donors has shown efficacy in multicenter settings. Hence, this approach could shorten the time for patients to receive VST therapy thus improving accessibility. In this review, we discuss the usage of VSTs for patients with primary immunodeficiency disorders in clinical trials, as well as future potential targets and methods to broaden the applicability of virus-directed T-cell immunotherapy for this vulnerable patient population.
Introduction
Primary immunodeficiency disorders (PID) are a growing group of congenital abnormalities of immunity, which range in severity from mild disorders of late childhood or adulthood, to profound, life-threatening disorders of infancy.1,2 Among patients with PID, infections are common and can be potentially life-threatening.3 In patients with poor to absent T-cell-mediated immunity, viral infections are often prolonged, and in some cases can be fulminant. In patients treated with hematopoietic stem cell transplantation (HSCT), presence of active infections has been well described to represent an independent risk factor for mortality.4 Newborn screening has enabled early detection of critical forms of PID such as severe combined immunodeficiency (SCID), which has been highly successful in enabling transplantation at an early age before exposure to multiple pathogens.5,6 However, current newborn screening using the TREC assay cannot identify most forms of combined immunodeficiency, many of which are also an indication for HSCT. Typically, older children and adult patients are referred for HSCT with a substantial preexisting burden of chronic infections.7
Prior natural history studies have generally identified herpesviruses such as cytomegalovirus (CMV) and Epstein-Barr virus (EBV), as well as respiratory viruses such as adenovirus as common causes of mortality both before and during HSCT.8-11 Though advances have been made in antiviral pharmacotherapy, available agents have significant toxicities, and rarely enable viral control without restoration of T-cell immunity.12,13 New antiviral agents such as letermovir have provided additional preventative measures,14 therapeutic options remain limited.
For more than 25 years, adoptive cellular therapy with virus-specific T cell (VST) has been used in the setting of HSCT for malignant and nonmalignant conditions with evidence of safety and efficacy.15-19 Unlike unmanipulated donor lymphocyte infusion, the risk of graft-versus-host disease (GVHD) is low when using VSTs.19 Beginning with studies targeting CMV and EBV using donor-derived T cells, current clinical trials have advanced to allow a broader range of targeted viruses, as well as an increased breadth of potential donor sources for VST production.20,21 Current methods of VST production use either selection of antigen-specific T cells using major histocompatibility complex multimers or cytokine capture technology, or ex vivo expansion following antigen stimulation.22-24 Selection technology has the advantage of same-day production and uses widely available platforms and kits, but requires an available donor who is immune to the targeted virus, and, generally, only a single virus may be targeted in each selection run. Ex vivo expansion involves stimulation of donor antigen presenting cells and coculture with lymphocytes, resulting in a relatively pure population of antigen-specific T cells. Current protocols typically use overlapping peptides pools as antigen sources, and can generate >108 VSTs in 10 to 12 days.11 Finally, expansion methods have permitted banking of “third-party” VSTs derived from healthy donors for use in the partially HLA-matched setting as an “off-the-shelf” therapeutic, which eliminates the cost and time required for customized patient-specific VSTs.25 This approach has shown success in recent studies with response rates of 74% to 95% after third-party VST administration.26-29
Here, we will review the use of VST therapy in patients with PID to date, as well as discussing the developing applications of virus-specific cellular therapy for this population.
Clinical use of VSTs in phase 1/2 studies
In 18 prior studies, 63 patients with PID have been treated with VST therapy (Table 1).17,20,22,23,28,30-41 Diagnoses vary, with SCID and hemophagocytic lymphohistiocytosis being most common (Figure 1). Cell doses vary widely, with studies dosing either by weight (between 0.8 × 102 and 2 × 106 / kg) or body surface area (5 ×106 to 1.35 ×108 VST/m2). Forty-four patients with PID received VSTs derived from their HSC donors, and 19 patients received VSTs from partially HLA-matched third-party donors (Figure 2). Thirteen patients received prophylactic VSTs after HSCT, with 11 patients remaining free of viral infection or reactivation for at least 1 year after HSCT and VST infusion. Two patients developed transient reactivations (1 with CMV, 1 with EBV), and in both patients the reactivation was controlled without the development of infection. The remaining 48 patients were treated for 1 to 3 active viral infections, with an overall response rate of 79%. In most studies, antiviral responses correlated with expansion of VSTs in the peripheral blood in the weeks after infusion.
Primary immunodeficiency patients in published studies of virus-specific T-cell therapy
| Study | Target | VST manufacturing method | Source | No. of PID patients treated | Results | AEs |
|---|---|---|---|---|---|---|
| Uhlin et al23 | CMV | Selection (A02:01 pp65/NLV pentamer) | Third party | 1 | PR, died after subsequent UCBT transplant | None |
| Bao et al30 | CMV | Culture, peptide | HSCT donor | 4 | CR in 2, PR in 1, NR in 1 | None |
| Creidy et al31 | CMV/Adv | Selection by IFN-γ secretion | HSCT donor | 5 | CR in 2, NR in 3 with death due to viral progression in 2 | DAH in 1, no GVHD |
| Feucht et al42 | Adv | Selection by IFN-γ secretion | HSCT donor | 1 | CR | None |
| Feuchtinger et al22 | Adv | Selection by IFN-γ secretion | HSCT donor | 7 | Responses in 21/30 patients overall (18 CR, 4 PR). No specific details regarding subjects with PID. | None |
| Leen et al17 | CMV, EBV, Adv | Culture, DC/LCL with Ad5f35-CMVpp65 vector | HSCT donor | 1 | CR (Adv) | None |
| Leen et al35 | EBV, Adv | Culture, DC/LCL with Ad5f35 vector | HSCT donor | 1 | Prophylactically treated; no subsequent viral infections | None |
| Papadopoulou et al20 | CMV, EBV, Adv, HHV6, BKV | Culture, peptide | HSCT donor | 4 | CR in 3, 1 patient with CR to HHV6 and EBV but NR to BKV | De novo grade 2 skin GVHD in 1 patient, resolved with topical therapy |
| Vickers et al, Wynn et al36,37 | EBV | Culture, LCL | Third party | 3 | CR in 1, progressive disease in 2 | GVHD grade 2 skin in 1 |
| Heslop et al38 | EBV | Culture, LCL | HSCT donor | 14 | CR in 4, PR in 3, NR in 1, 5 treated prophylactically without reactivation | None |
| Doubrovina et al19 | EBV | Culture, LCL | HSCT donor or third party | 3 | CR in 1 (third party), NR in 2 | None |
| Naik et al43 | CMV, EBV, Adv | Culture (LCL, peptide, or viral vector) | HSCT donor or third party | 13 (newly reported) | CR in 6, PR in 3, NR in 2, 2 free of viral infections | aGVHD in 3 (grade 2-3), cGVHD in 1 (grade 1) |
| Miller et al39 | Adv | Culture, peptide | Third party | 1 | CR | None |
| Punwani et al40 | CMV | Culture, peptide | HSCT donor | 1 | CR | None |
| Withers et al28 | CMV, EBV, Adv | Culture, peptide | Third-party | 4 | CR in 3, NR in 1 | None |
| Gerdemann et al34 | CMV, EBV, Adv | Culture, plasmid nucleofection | HSCT donor | 1 | CR | None |
| Tzannou et al29 | CMV, EBV, Adv, HHV6, BKV | Culture, peptide | Third party | 2 | CR (Adv) in 2, PR (CMV) in 1 | Flare of preexisting GI GVHD in 1 (in setting of IS wean); resolved with therapy |
| Abraham et al41 | CMV, EBV, Adv | Culture (DC, LCL with Ad5f35-pp65 or peptide) | HSCT donor (UCBT) | 3 | All free of viral infection | None |
| Tzannou et al44 | CMV | Culture, peptide | Third party | 2 | CR in 2 | None |
| Study | Target | VST manufacturing method | Source | No. of PID patients treated | Results | AEs |
|---|---|---|---|---|---|---|
| Uhlin et al23 | CMV | Selection (A02:01 pp65/NLV pentamer) | Third party | 1 | PR, died after subsequent UCBT transplant | None |
| Bao et al30 | CMV | Culture, peptide | HSCT donor | 4 | CR in 2, PR in 1, NR in 1 | None |
| Creidy et al31 | CMV/Adv | Selection by IFN-γ secretion | HSCT donor | 5 | CR in 2, NR in 3 with death due to viral progression in 2 | DAH in 1, no GVHD |
| Feucht et al42 | Adv | Selection by IFN-γ secretion | HSCT donor | 1 | CR | None |
| Feuchtinger et al22 | Adv | Selection by IFN-γ secretion | HSCT donor | 7 | Responses in 21/30 patients overall (18 CR, 4 PR). No specific details regarding subjects with PID. | None |
| Leen et al17 | CMV, EBV, Adv | Culture, DC/LCL with Ad5f35-CMVpp65 vector | HSCT donor | 1 | CR (Adv) | None |
| Leen et al35 | EBV, Adv | Culture, DC/LCL with Ad5f35 vector | HSCT donor | 1 | Prophylactically treated; no subsequent viral infections | None |
| Papadopoulou et al20 | CMV, EBV, Adv, HHV6, BKV | Culture, peptide | HSCT donor | 4 | CR in 3, 1 patient with CR to HHV6 and EBV but NR to BKV | De novo grade 2 skin GVHD in 1 patient, resolved with topical therapy |
| Vickers et al, Wynn et al36,37 | EBV | Culture, LCL | Third party | 3 | CR in 1, progressive disease in 2 | GVHD grade 2 skin in 1 |
| Heslop et al38 | EBV | Culture, LCL | HSCT donor | 14 | CR in 4, PR in 3, NR in 1, 5 treated prophylactically without reactivation | None |
| Doubrovina et al19 | EBV | Culture, LCL | HSCT donor or third party | 3 | CR in 1 (third party), NR in 2 | None |
| Naik et al43 | CMV, EBV, Adv | Culture (LCL, peptide, or viral vector) | HSCT donor or third party | 13 (newly reported) | CR in 6, PR in 3, NR in 2, 2 free of viral infections | aGVHD in 3 (grade 2-3), cGVHD in 1 (grade 1) |
| Miller et al39 | Adv | Culture, peptide | Third party | 1 | CR | None |
| Punwani et al40 | CMV | Culture, peptide | HSCT donor | 1 | CR | None |
| Withers et al28 | CMV, EBV, Adv | Culture, peptide | Third-party | 4 | CR in 3, NR in 1 | None |
| Gerdemann et al34 | CMV, EBV, Adv | Culture, plasmid nucleofection | HSCT donor | 1 | CR | None |
| Tzannou et al29 | CMV, EBV, Adv, HHV6, BKV | Culture, peptide | Third party | 2 | CR (Adv) in 2, PR (CMV) in 1 | Flare of preexisting GI GVHD in 1 (in setting of IS wean); resolved with therapy |
| Abraham et al41 | CMV, EBV, Adv | Culture (DC, LCL with Ad5f35-pp65 or peptide) | HSCT donor (UCBT) | 3 | All free of viral infection | None |
| Tzannou et al44 | CMV | Culture, peptide | Third party | 2 | CR in 2 | None |
Adv, adenovirus; AE, adverse event; aGVHD, acute GVHD; cGVHD, chronic GVHD; DAH, diffuse alveolar hemorrhage; DC, dendritic cell; IS, immunosuppression; LCL, lymphoblastoid cell line; UCBT, umbilical cord blood transplant.
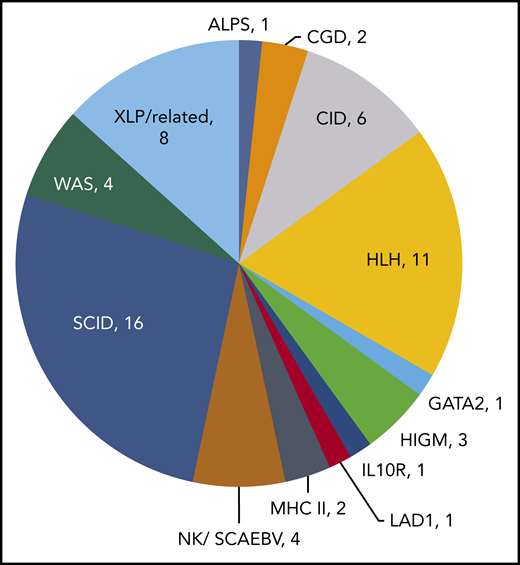
Diagnoses of primary immunodeficiency patients who were treated with virus-specific T-cell therapy. ALPS, autoimmune lymphoproliferative syndrome; CGD, chronic granulomatous disease; CID, combined immunodeficiency; HLH, hemophagocytic lymphohistiocytosis; HIGM, hyper-immunoglobulin M syndrome; LAD1, leukocyte adhesion deficiency type I; MHC II, major histocompatibility complex deficiency type II; NK, natural killer; SCAEBV, severe chronic active EBV; WAS, Wiskott-Aldrich syndrome; XLP, X-linked lymphoproliferative disorder.
Diagnoses of primary immunodeficiency patients who were treated with virus-specific T-cell therapy. ALPS, autoimmune lymphoproliferative syndrome; CGD, chronic granulomatous disease; CID, combined immunodeficiency; HLH, hemophagocytic lymphohistiocytosis; HIGM, hyper-immunoglobulin M syndrome; LAD1, leukocyte adhesion deficiency type I; MHC II, major histocompatibility complex deficiency type II; NK, natural killer; SCAEBV, severe chronic active EBV; WAS, Wiskott-Aldrich syndrome; XLP, X-linked lymphoproliferative disorder.

Targeting CMV
CMV is a beta herpesvirus that establishes lifelong latency in monocytes.45 Both acute and reactivation infections can result in multisystem disease, including pneumonitis, hepatitis, and encephalitis. Though antiviral medications including ganciclovir, foscarnet, and newer agents such as brincidofovir and letermovir have reduced infections rates in transplant recipients, they are expensive and often complicated by both toxicity and antiviral resistance.8,14,46 Additionally, although most newer agents have been successful for CMV prophylaxis, they have limited data as therapy for viral disease, and accordingly may be less useful for patients with CMV disease that precedes HSCT. Vaccine strategies currently have limited value in patients with PID, and presently there are no approved vaccines targeting CMV.47 CMV has been treated with VST therapy in multiple prior studies, in which either ex vivo expansion or selection methods were used.17,23,30,31,33,44 In total, 20 patients with PID were treated with VSTs for CMV infection. Thirteen patients received VSTs derived from HSCT donors, with 8 patients having antiviral responses (5 with resolution`` and 3 with partial responses; Figure 3). Seven patients were treated for CMV with VSTs from third-party donors, with resolution of viremia in 5, and partial response (sustained decrease in viral load without resolution) in 2 of those treated (Figure 4). Overall, 14 of the 20 patients received VSTs produced by ex vivo expansion, with antiviral responses in 12, and 6 patients received VSTs produced via selection approaches, with responses in 2 patients. Responses occurred in patients with pneumonitis, retinitis, and encephalitis, suggesting that VSTs (irrespective of the manufacturing strategy used) can effectively traffic to sites of infections.
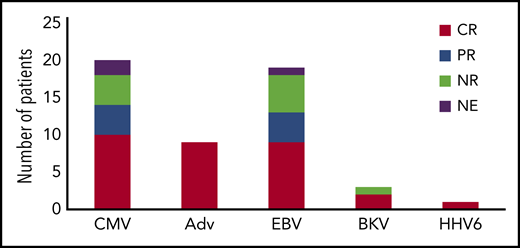
Antiviral responses following VST therapy by virus. CR, complete response (resolution of viremia and associated symptoms); PR, partial response (sustained >50% decrease in viral load); NE, not evaluable; NR, no response.
Antiviral responses following VST therapy by virus. CR, complete response (resolution of viremia and associated symptoms); PR, partial response (sustained >50% decrease in viral load); NE, not evaluable; NR, no response.

Targeting of EBV
EBV is a gamma herpesvirus that establishes lifelong latency in B cells, and, in immunocompromised patients, can cause fulminant infectious mononucleosis or lymphoproliferative disease.10,48 Though use of rituximab has reduced the incidence of posttransplant lymphoproliferative disease, patients with PID remain at risk, and chronic EBV viremia occurs in patients with many forms of PID even before HSCT. Newer monoclonal antibody therapies such as obinutuzumab and daratumumab have been successful in cancer trials,49,50 but have limited data for treatment of posttransplant lymphoproliferative disease (PTLD). In prior VST studies, EBV has been one of the most common targets, and, in the published literature, 31 patients with PID have received VSTs targeting EBV either therapeutically or prophylactically.19,33,36-38 Of 12 patients who received donor-derived VSTs targeting EBV (either as a single target or alongside other viruses) as prophylaxis post HSCT, only 1 patient developed a subsequent EBV reactivation, which resolved without the need for rituximab. Of 19 patients who were treated for active EBV, 9 had complete resolution of disease and 4 achieved a partial response with sustained reduction in EBV viral load albeit without complete clearance. Antiviral responses occurred in 7 of 11 patients who received donor-derived VSTs; 4 of 8 patients treated with third-party derived VSTs. One patient was successfully treated for central nervous system lymphoproliferative disease, again supporting that VSTs can traffic to sites of infection including the brain.
Targeting of adenovirus
Adenovirus is a nonencapsulated DNA virus and a leading cause of respiratory and gastrointestinal (GI) disease in immunocompromised patients. Cidofovir has activity against adenovirus, but usage is often limited by renal toxicity. Brincidofovir has shown efficacy against adenovirus in small studies without significant renal toxicities, but has been associated with GI toxicities.51,52 Typically, treatment with cidofovir or brincidofovir has been associated with incomplete viral clearance in patients with PID.53,54 In published studies, 9 patients received VST therapy for adenoviremia.17,31,33,55 Four patients received VSTs derived from stem cell donors (2 using selection and 2 using ex vivo expansion methodologies). Additionally, 5 patients received VSTs derived from third-party donors (all generated using expansion approaches). Eight of the 9 patients cleared the adenoviral infection, supporting the efficacy of this approach irrespective of the VST manufacturing strategy used. Eight patients received adenovirus-specific VSTs as prophylaxis, and none developed adenoviral infections during their follow-up period (maximum, 1 year).
Targeting of other viruses
Papadopolou et al described the use of donor-derived VSTs targeting 5 viruses: CMV, EBV, adenovirus, BK virus, and human herpesvirus 6 (HHV6).20 BK virus is a polyomavirus associated with hemorrhagic cystitis and rare cases of pervasive multifocal leukoencephalopathy.56,57 HHV6B is the cause of roseola and can cause encephalitis in immunocompromised patients.58 In this study, 4 patients with PID disorders were treated with VSTs targeting these 5 viruses after HSCT. Of these 4 patients, 3 received VSTs for BK viremia, and the fourth patient had HHV6 reactivation. The BK viremia resolved in 2 of the 3 patients treated for BK. The nonresponding patient was subsequently found to have a donor who was BKV seronegative. The 1 patient who received VSTs for HHV6 had a complete response. In a follow-up study, this group targeted the same 5 viruses using third-party donor off-the-shelf VSTs, and 2 patients with PID were treated for active viral infections (adenovirus in a patient with SCID; adenovirus and CMV in a patient with chronic granulomatous disease).29 The patient with SCID cleared the adenovirus infection after receiving the third-party VSTs. After VST infusion, the patient with chronic granulomatous disease demonstrated improvements in CMV and adenovirus DNA levels, but the patient ultimately succumbed to fungal pneumonia.
Production techniques and clinical use of VSTs derived from naïve T cells
The majority of prior VST studies have depended on donors with preexisting immunity to targeted viruses. However, HSCT from umbilical cord blood and donors who are CMV or EBV seronegative represents the highest risk setting for viral infections and disease because of the lack of memory T cells.59 It has been demonstrated that VSTs can be expanded from the 20% fraction of umbilical cord blood units, as well as CMV-naïve adult donors, through the use of naïve T-cell isolation and serial stimulations with donor-derived antigen-presenting cells, including dendritic cells and lymphoblastoid cells lines. These protocols require prolonged culture periods (median, 7 weeks).21,60 Nonetheless, 14 patients have been treated with cord blood-derived VSTs following cord blood transplantation, including 3 with SCID.41 All 3 of these patients remained free of the targeted viruses (CMV, EBV, adenovirus), in spite of high risk of viral complications. Thus, use of VSTs derived from virus-naïve donors is possible and likely beneficial, and simplification of production protocols will improve the feasibility of this treatment approach.61
Persistence of infused VSTs in vivo
The fate of infused VSTs and how they contribute to antiviral immune reconstitution has been a key question in prior studies.11,25 It has been observed that reduction in viral loads often correlates with expansion of T cells that recognize targeted viral antigens.17,20,62 When using donor-derived VSTs in the transplant setting, the source of this T-cell expansion can be challenging to determine. Early studies administering EBV-specific T cells used cells that were gene marked using a retroviral vector expressing a neomycin resistance gene, which allowed tracking of the infused cells.38 The gene-marked T cells were detectable via polymerase chain reaction for up to 105 months postinfusion, and included both CD4+ and CD8+ memory T cells that readily expanded in response to restimulation with EBV antigens. Similarly, tracking of infused VSTs via T-cell receptor (TCR) sequencing has demonstrated persistence of infused cells for >3 years and demonstrated that clonotypes associated with infused VSTs dynamically expand and contract in tandem with viral loads, representing between 0.6% and 55% of peripheral T-cell clonotypes.63 In the setting of third-party T-cell therapy, persistence is more variable. Several studies using short tandem repeat analysis demonstrated persistence of infused VSTs for up to 3 months,23,26 after which it is theorized that emerging allografts will reject the partially HLA-matched cells. Neuenhahn et al reported no persistence in 7 of 8 patients treated with Streptamer-isolated CMV-specific T cells derived from third-party donors.64 However, low cell dose, lack of CD4+ help, and use of corticosteroids may have interfered with VST engraftment in these patients. Usage of high-dose corticosteroids or lymphotoxic agents including alemtuzumab are well-described to interfere with VST engraftment. Thus, many factors could influence engraftment and subsequently the efficacy of third-party VSTs in vivo.
Safety of VSTs
GVHD
GVHD is a primary concern with the use of adoptively transferred allogeneic T cells, especially in the third-party setting, but unlike donor lymphocyte infusion, rates of GVHD in all VST trials have been relatively low.19 Most prior trials have performed testing of VST products for alloreactivity via 51Cr release testing, though a previous retrospective analysis suggested that GVHD rates are low even with evidence of alloreactivity in vitro.65 When administering VSTs derived from a patient’s own transplant donor, reported GVHD rates are extremely low (<11%).20,34,35,62 For example, Naik et al described GVHD occurring in only 3 of 29 patients with PID receiving donor-derived VSTs.43 Most importantly, all occurrences of GVHD were only grade 1 or 2, and all patients were therapy responsive.33 In the third-party setting, rates of reported GVHD are similarly rare, occurring in only 3 of 17 patients with PID.28,31,32 Of these 3 reports, 2 were de novo (grade 2 skin or GI disease), and the other was a flare of GI GVHD occurring in the setting of an immunosuppression wean. However, most published studies also lack control groups, making it impossible to know whether GVHD arose from VST infusion or from the HSCT per se.
CRS
Cytokine release syndrome (CRS) is a multisystem inflammatory disorder resulting from the robust proliferation and nonantigen specific activity of immune effector cells.66,67 Primary mediators include interferon-γ (IFN-γ), interleukin-6 (IL-6), IL-1, and tumor necrosis factor-α (TNF-α). Symptoms may include fever, tachycardia, hypotension, and encephalopathy. The clinical spectrum of CRS is wide, with milder cases often referred to as “engraftment syndrome,” and more serious cases being indistinguishable from hemophagocytic lymphohistiocytosis.
CRS is well-described with the use of CD19 chimeric antigen receptor (CAR) T cells targeting B-cell malignancies, with reported CRS rates of any grade ranging from 37% to 93% in patients with non-Hodgkin lymphoma and 77% to 93% in patients with acute or chronic leukemia.68,69 Though far less common when using genetically unmodified VSTs, CRS has been described in a minority (<2%) of patients after infusion. In the rare instances when CRS does occur post-VST infusion, it is typically observed in the setting of bulky EBV lymphoproliferative disease, or disseminated viral disease with high viral loads.11 Given their elevated risk of disseminated viral disease, PID patients may be at slightly higher risk of CRS, though it has not been reported in previous cases. An added challenge with the use of VSTs is that CRS may be nearly indistinguishable from progressive viral disease. However, longitudinal monitoring of cytokines and inflammatory markers alongside viral loads may aid in increasing the accuracy of the diagnosis.
Anticytokine therapies including tocilizumab and TNF antagonists have demonstrated efficacy against CRS.70 Severe cases may require corticosteroids, but as this will inactivate VSTs, they should be avoided unless absolutely necessary.
Special situations: VST therapy in the pre-HSCT period
Though the vast majority of recipients of VST therapy have been treated in the post-HSCT setting, viral infections can be potentially life-threatening before transplant in patients with severe T-cell deficiency, and the presence of active infection has been shown to worsen survival rates post-HSCT.3,4 To date, 6 patients with PID have been reported to have received VST therapy before HSCT. Wynn et al described a child with EBV lymphoproliferative syndrome (later confirmed to have CTPS1 deficiency), who received EBV-specific T cells before cord blood transplant, followed by several additional VST doses, resulting in a complete resolution of the lymphopoliferative disease lesions without toxicity.37 Miller et al described the treatment of a baby with RAG1 SCID who received 2 doses of VSTs for disseminated adenovirus before matched unrelated donor HSCT, with subsequent clearance of adenovirus also without toxicity.39 In most successful cases, after VST infusion patients proceeded to transplant expeditiously, and it is unclear if tandem graft infusion also facilitated VST engraftment. As some forms of PID are also associated with defects in antigen-presenting cells, restoration of these subsets may be necessary to facilitate VST activity, though whether a certain degree of donor chimerism may aid VST activity is also unknown. Successful VST therapy before HSCT has required multiple doses in all cases, which could suggest either a higher cell dosing requirement, or limited persistence in this population. Thus, the ideal cell dose and frequency of reinfusion still remain unclear. Graft rejection after pretransplant VST therapy has not been noted in any cases, but to date, the number of patients treated in this setting is very small. It also remains to be seen if VSTs may be effective for the treatment of viral infections in PID with immunodysregulatory features, in which autoreactive T cells could potentially reject VSTs. Thus, although VST therapy may be a useful adjunctive therapy in this period, larger trials are ongoing to better address these questions (NCT02510404, NCT03475212).
Limitations of VST therapy
Despite the reported successes of VST therapy, there remain many obstacles to widespread utilization. Few controlled VST studies have been performed, and response definitions have varied between studies, making comparison of VST and pharmacotherapy studies difficult. Production of VSTs currently requires a clean room or good manufacturing practice facility and the regulatory infrastructure to support investigsational new drug (IND)-based studies.71 Use of commercial selection platforms reduces these requirements to some degree, though production costs are high when using selection kits. When targeting multiple viruses in a single VST product, antigenic competition has been a concern, where responses against immunodominant antigens such as CMV-pp65 may skew the T-cell population and reduce responses to other viral antigens.72 To date, this has not limited the ability to treat patients with VSTs targeting up to 5 different viruses in a single product.20,29
With exception of early studies in which VSTs were gene-marked,38 VSTs used clinically have not been genetically modified, and are prone to inactivation by immunosuppressive medications. Because GVHD is a frequent comorbidity in recipients of HSCT, many forms of PID have immunodysregulatory manifestations that also require immunosuppressive therapy. This creates the problem of having to treat viral infections while also suppressing autoreactive T cells. Previous studies suggest that drugs such as calcineurin inhibitors and mycophenolate do not appreciably impair VST activity in vivo.73,74 However, corticosteroid therapy at moderate to high doses (typically >0.5 mg/kg/day) will inactivate VSTs, and published reports suggest that higher dose steroid use is associated with a poor clinical response corresponding to a lack of VST expansion in vivo.28,31 The effect of newer biologic therapies used to prevent and treat GVHD such as monoclonal antibodies targeting cytokines and JAK inhibitors on VSTs is not well studied. TNF-α blockade, commonly used to treat inflammatory conditions, does not appear to be critical for antiviral immunity, nor does it seem to blunt VST activity in vivo.75 IL-6 blockade using tocilizumab, which is a first-line therapy for cytokine release syndrome, inhibits Th17 immunity and may dampen, but does not inactivate, T-cell activity.76 JAK inhibitors are potent suppressors of interleukin and interferon signaling, and viral reactivation has been a complication in patients receiving this therapy.77,78 It is accordingly important to account for the class and dose of immunosuppression used in patients who are being considered for VST therapy.
In the setting of third-party VST therapy, matching algorithms for selecting partially HLA-matched VST products for a given recipient remain entirely dependent on expert consensus. Mapping of antiviral HLA restrictions has improved the response rates in recent clinical studies over earlier trials that treated based on best overall HLA match.26-29 For viruses such as CMV, several key T-cell epitopes are known to be immunodominant (such as the A02:01 restricted NLV and B07:02 restricted TPR and RPH epitopes within pp65) and seem to correlate with successful antiviral responses in vivo. However, responses to immunodominant epitopes tend to crowd out responses to minor epitopes, and a better understanding is developing of the immune hierarchy for many viral antigens. The degree of conservation of epitopes in circulating viruses also contributes to the chance of successful response to VST therapy because escape mutations have been described in EBV.79 Though the response rate for reported adenovirus patients has been encouraging and likely reflects a strong degree of cross-reactivity between T-cell epitopes in circulating adenovirus genotypes, adenovirus remains a major problem in the PID population, and additional study evaluating immunodominant epitopes for ideal VST matching is needed.
Potential future targets for VST therapy
Though prior studies have successfully targeted key viruses such as CMV and adenovirus, many other pathogens are a risk to patients with PID. Varicella-zoster virus has been targeted prophylactically in 1 prior VST trial, but not used therapeutically.80 Other preclinical viral targets include herpes simplex virus, respiratory syncytial virus, human metapneumovirus, human parainfluenza virus-3, and human papillomaviruses (Table 2).32,81-83 T-cell therapy targeting enteric viruses such as norovirus may also be beneficial, and immunodominant T-cell antigens have recently been described (Hanajiri et al). Beyond viruses, Mucorales and mycobacterial species may also be future targets for adoptive T-cell therapy.84-86
Future targets for VST therapy
| Potential VST targets | Targets/antigen sources | Clinical studies | Reference |
|---|---|---|---|
| HSV | A02-restricted T cells expanded | None | Ma et al82 |
| VZV | VZV vaccine | Used in prophylaxis post-BMT | Ma et al80 |
| HPV | E6, E7 peptide libraries | Prior use against HPV-associated cancers | McCormack et al83 |
| Respiratory viruses | |||
| Influenza A | Nucleocapsid protein, Matrix protein 1 peptide libraries | None | Vasileiou et al87 |
| Respiratory syncytial virus | Nucleoprotein and glycoprotein F0 protein peptide libraries | None | Vasileiou et al87 |
| Human metapneumovirus | Nucleocapsid, fusion protein peptide libraries | None | Tzannou et al32 |
| Human parainfluenza virus | HPIV3 matrix protein | None published/NCT03180216 | McLaughlin et al81 |
| Enteric viruses | |||
| Norovirus | VP1, NS6 peptide libraries | None | Hanajiri et al |
| Mycobacteria spp. | Ag85B, PPE68, ESAT6, CFP10 peptide libraries (Mycobacteria tuberculosis) | None | Patel et al85 |
| Mucormycosis | Rhizopus oryzae extract | None | Castillo et al86 |
| Potential VST targets | Targets/antigen sources | Clinical studies | Reference |
|---|---|---|---|
| HSV | A02-restricted T cells expanded | None | Ma et al82 |
| VZV | VZV vaccine | Used in prophylaxis post-BMT | Ma et al80 |
| HPV | E6, E7 peptide libraries | Prior use against HPV-associated cancers | McCormack et al83 |
| Respiratory viruses | |||
| Influenza A | Nucleocapsid protein, Matrix protein 1 peptide libraries | None | Vasileiou et al87 |
| Respiratory syncytial virus | Nucleoprotein and glycoprotein F0 protein peptide libraries | None | Vasileiou et al87 |
| Human metapneumovirus | Nucleocapsid, fusion protein peptide libraries | None | Tzannou et al32 |
| Human parainfluenza virus | HPIV3 matrix protein | None published/NCT03180216 | McLaughlin et al81 |
| Enteric viruses | |||
| Norovirus | VP1, NS6 peptide libraries | None | Hanajiri et al |
| Mycobacteria spp. | Ag85B, PPE68, ESAT6, CFP10 peptide libraries (Mycobacteria tuberculosis) | None | Patel et al85 |
| Mucormycosis | Rhizopus oryzae extract | None | Castillo et al86 |
BMT, bone marrow transplantation; HPV, human papillomavirus; VZV, varicella-zoster virus.
Future of VST and related cellular therapies
In light of the decades of phase 1 and 2 data on antiviral T-cell therapy and the recent US Food and Drug Administration approval of CD19 CAR T-cell therapy products, it is probable that antiviral cellular therapy may be approved in the near future. Use of regional VST banks (academic and commercial) will be essential for broadening accessibility to this therapy. The expanding use of CAR-T therapies is likely to hasten this path to commercialization, as the infrastructure requirements for CAR-T and VST therapy are similar. Future trials will also expand T-cell therapies for patients with PID to target additional pathogens, both therapeutically and prophylactically. Genetic modification of antigen-specific T cells may allow VST treatment of infections in the setting of immunosuppression for GVHD or other inflammatory diseases. Prior studies have demonstrated the feasibility of modification of glucocorticoid receptors and cyclophilin-binding proteins to allow tolerance of glucocorticoids and calcineurin inhibitors, respectively.88,89 Reversal of T-cell exhaustion via checkpoint inhibitors via targeting the PD1-PDL1 pathway has also been successful in restoring antiviral T-cell function in recent studies,90,91 and may have a role in treatment of some infectious diseases, either alone or in combination with adoptive T-cell therapy. Finally, as more data are collected regarding the breadth of TCRs that target specific pathogens, TCR libraries may allow future studies in which antigen specificity is bestowed on effector T cells via transduction of paired TCR-α/β chains.
Summary
Virus-specific T cells and related cellular therapies have shown great promise in combating refractory viral infections in patients with PID, and may be expanded to the treatment of other opportunistic infections. To date, the efficacy of VST therapy in PID patients appears to comparable to other patient populations following HSCT. However, the heterogeneity of PID disorders warrants further studies to determine the efficacy and unique safety aspects of cellular therapies in this population. Though access has been limited to date because of the degree of customization and regulatory requirements inherent in this treatment, process simplification and commercialization will expand accessibility to more patients, and hopefully with time, render virus-associated mortality (especially after HSCT) a thing of the past.
Acknowledgments
This work was supported by funding from the National Institutes of Health, National Heart, Lung, and Blood Institute (K23-HL136783-01) (M.D.K.), the Board of Visitors of Children’s National Medical Center, and the Jeffrey Modell Foundation.
Authorship
Contribution: M.D.K. and C.M.B. designed the research, analyzed data, and wrote the paper.
Conflict-of-interest disclosure: M.D.K. is on a scientific advisory panel for Gilead Sciences. C.M.B. is on the scientific advisory boards for Cellectis, has stock options in Neximmune and Torque Therapeutics, and has stock or ownership in Mana Therapeutics. C.M.B. and M.D.K. have filed patent applications related to the subject of this paper.
Correspondence: Michael D. Keller, Division of Allergy and Immunology, Children’s National Health System, 111 Michigan Ave NW, Washington DC 20010; e-mail: mkeller@childrensnational.org.

This feature is available to Subscribers Only
Sign In or Create an Account Close Modal