


Abstract
Epstein-Barr virus (EBV) is an enigma; on one hand, it infects and persists in latent form in the vast majority of the global population, causing relatively benign disease in otherwise healthy individuals. On the other hand, EBV represents the first identified oncogenic virus, capable of causing ≥7 different types of malignancies, usually in immunocompromised individuals. Furthermore, some individuals with defined inborn errors of immunity exhibit extreme susceptibility to EBV-induced disease, developing severe and often fatal infectious mononucleosis, hemophagocytic lymphohistiocytosis, lymphoproliferative disease, and/or EBV+ B-cell lymphoma. Thus, host and pathogen have coevolved to enable viral persistence and survival with minimal collateral damage to the healthy host. However, acquired or genetic disruptions to host defense that tip the balance in favor of EBV can have catastrophic effects. The study of primary immunodeficiencies has provided opportunities to define nonredundant requirements for host defense against EBV infection. This has not only revealed mechanisms underlying EBV-induced disease in these primary immunodeficiencies but also identified molecules and pathways that could be targeted to enhance the efficacy of an EBV-specific vaccine or treat severe EBV infection and pathological consequences in immunodeficient hosts.
Introduction
Epstein-Barr virus (EBV; human herpesvirus 4 [HHV4]) belongs to the human herpesvirus family, which also includes HSV1, HSV2, varicella zoster virus (VZV), cytomegalovirus (CMV), and Kaposi sarcoma virus, which can all establish lifelong infection in humans.1 Indeed, >90% of the world’s population has been exposed to, and thus carries, EBV.1-4 The clinical consequences of primary EBV infection depend on the age and immunocompetent state of the host. Thus, while EBV infection in children is mostly asymptomatic, ∼10% of adolescents develop infectious mononucleosis (IM).2-4 Importantly, EBV is oncogenic, with numerous cancers (B, T, and natural killer [NK] cell lymphoma; Hodgkin lymphoma; smooth muscle tumors; and nasopharyngeal and gastric carcinomas) being associated with EBV infection.2,4-6
The importance of robust immunity against EBV is apparent from severe outcomes of infection in immunocompromised individuals; for example, transplant recipients frequently develop lymphoproliferative disease due to EBV-induced transformation of donor B cells.2,5,6 More strikingly is the dramatic and often-fatal outcomes of EBV infection (severe IM, lymphoproliferation, hemophagocytic lymphohistiocytosis [HLH], and lymphoma) in individuals with primary immunodeficiencies (PIDs).5,7-9 These clinical observations indicate that ongoing immune surveillance is necessary to maintain virus-host homeostasis and unequivocally established the key role of the immune system in preventing some hematological malignancies. These findings also highlighted the pathogenicity of EBV and the need for developing an effective vaccine.
Regulation of EBV infection by innate and adaptive immunity
The innate and adaptive arms of the immune system mediate host response to EBV infection. NK and NKT cells restrain EBV-mediated B-cell transformation in vitro,10-13 and EBV-induced pathology is more severe in NK-depleted humanized mice than in NK-sufficient murine hosts.12,14 Thus, innate immunity provided by NK and NKT cells likely contribute to initial control of EBV infection in humans.
Adaptive immunity is fundamental for protection against EBV.6 This is clear from the frequency of life-threatening EBV-induced disease in individuals with defects in T-cell development or function.6,9,15 During primary EBV infection in immunocompetent individuals, peripheral blood T cell numbers increase dramatically, with EBV-specific cells comprising up to 50% of peripheral CD8+ and ∼1% of CD4+ T cells, contracting to ∼2% to 5% and ∼0.1%, respectively, of memory CD8+ and CD4+ T cells upon resolution of infection.4,6,16,17 Elevated serum inflammatory (interferon γ [IFN-γ], tumor necrosis factor α [TNF-α], and interleukin-6 [IL-6]) and immunoregulatory (IL-10 and transforming growth factor β) cytokines coincide with T-cell expansion and acquisition of cytotoxic function during acute infection.4,6,17-21 It is not clear whether EBV-specific antibodies (Abs) contribute to controlling viral infection.4,6 Abs induced following vaccination reduced the severity of EBV infection.22 However, these Abs did not prevent infection, indicating Abs were insufficient for host defense.22
Insights from PIDs
PIDs are caused by monogenic germline mutations that disrupt the development, differentiation, and/or effector function of immune cells. Thus, affected individuals are highly susceptible to severe and often-fatal infections.15 This can be either to a broad or very narrow range of infections, highlighting the specialization of the adaptive immune response.9,15
Through the application of next-generation sequencing, the rate of discovery of inborn errors of immunity has increased rapidly. More than 400 genetic causes of immune dysregulation have been identified,15 with ∼40% discovered over the past ∼5 years by unbiased DNA-sequencing approaches.23 Several PIDs have been described in which chronic EBV viremia and/or EBV-induced disease (fulminant IM, HLH, lymphoproliferation, lymphoma, and dysgammaglobulinemia) are common clinical features of affected individuals.7-9,15,24,25 Studies of these patients have provided remarkable insight into the unique requirements for efficacious immune responses against EBV. Here, we review PIDs that have identified pathways necessary for host defense against EBV and mechanisms underlying disease pathogenesis following EBV infection (Table 1).
Human inborn errors of immunity resulting in severe EBV-induced disease
| Mutated gene | Clinical features | Viral infections | Outcomes | Cellular phenotype | Treatment | Mechanism of disease | References |
|---|---|---|---|---|---|---|---|
| SH2D1A (SAP) (n > 100) | Age of onset, 0.5-40 y EBV viremia (75%), FIM/HLH (∼50-60%), hypogammaglobulinemia (40-50%), B lymphoma (Burkitt/NHL; EBV+ and EBV−; ∼25%) | EBV | ∼30-80% mortality | ↓ memory B cells, no NKT cells, ↑ TEMRA (exhausted-type) CD8+ T cells | IV Ig, B-cell depletion (rituximab), chemotherapy for lymphoma, HSCT (curative in >90% of cases) | ↓SLAM/SAP-dependent activation of NK and Ag-specific CD8+ T cells by Ag-presenting or transformed B cells | 26-34 |
| ITK (n = 22) | Age of onset, 3-44 y EBV viremia (100%), EBV-induced lymphoproliferation (∼70%), EBV+ B-cell lymphoma (mostly Hodgkin; 1 NHL, 1 Burkitt lymphoma) (∼80%), other infections (epidermodysplasia verruciformis; 2 patients), CD4+ T-cell lymphopenia (∼65%), splenomegaly (∼60%), progressive hypogammaglobulinemia (>50%), some autoimmunity (25%) | EBV,CMV, VZV,HPV | 45% mortality | Normal % CD3+ T cells but ↓ naive CD4+ T cells, ↓ NKT cells | Rituximab, chemotherapy for lymphoma, IV Ig (not particularly effective), HSCT (5/7 alive and well post-HSCT) | ↓T cell activation (↓PLCγ activation) | 35-47 |
| MAGT1 (n = 22) | Age of onset, 3-45 y, chronic EBV viremia (>90%), EBV-induced lymphoproliferation (not HLH), EBV+ B lymphoma (Burkitt/Hodgkin/DLBCL; 70%), CD4+ T-cell lymphopenia (∼50%), splenomegaly, progressive hypogammaglobulinemia (>50%), intellectual/developmental disability (2/3 patients), autoimmunity (cytopenia, Guillain-Barre syndrome, hepatitis), recurrent infections (80%), Kaposi sarcoma (1 patient), Castleman disease (1 patient) | EBV,VZV, HSV, CMV, KSHV; molluscum contagiosum | ∼25% mortality | ↑ B cells, ↓ memory B cells, inverted CD4/CD8 ratio, ↓ NKG2D expression on NK and CD8+ T cells | Rituximab, R-CHOP; IV Ig; HSCT (2/4 patients died post HSCT); risk of hemorrhage pre-/post-HSCT; Mg supplementation (though variable effect) | ↓T cell activation (↓ TCR-induced Mg2+/Ca2+ flux, PLCγ activation); ↓ NKG2D impairs NK/CD8+ T cells cytotoxicity toward B cells; glycosylation defect (likely impairs expression of NKG2D, and other important immunoregulatory glycoproteins, as well as potentially affecting stability of serum Ig) | 46-56 |
| CTPS1 (n = 18) | Age of onset: 0-5 y EBV viremia (> 90%) EBV-lymphoproliferative diseases (30-50%) Severe IM (∼50%) Recurrent viral/bacterial infections (100%) Poor Ab responses to vaccines/infections; hypogammaglobulinemia (∼30%) | EBV,VZV (∼50%), HSV, CMV, HHV6 (∼20%); norovirus, adenovirus | ∼40% mortality | ↓ NKT, NK, MAIT cells; ↓ memory B and CD8+ T cells | Rituximab, adoptive EBV-specific CD8+ T cell therapy, IV Ig, HSCT (11/14 alive and well) | ↓ activation/proliferation of T and B cells (↓ de novo pyrimidine synthesis), CD27/CD70-induced T cell proliferation may be impaired, ↓IFN-γ production | 57-60 |
| RASGRP1 (n = 9) | Age of onset, 0-7 y EBV viremia (>70%) EBV+ B-lymphoma (Hodgkin), smooth muscle tumor (∼70%) CD4+ T cell lymphopenia Poor Ab responses to some vaccines/infections (∼20%) Epidermodysplasia verruciformis (1 patient) Lymphadenopathy Autoimmunity (cytopenias, ANA, hepatitis; ∼50%) Recurrent bacterial/viral infections (pneumonia) (100%) HLH (1 patient) | EBV,CMV, VZV, HSV1,HPV;molluscum contagiosum | ∼30-35% mortality (3/9 patients deceased) | ↓ naive CD4+ and CD8+ T cells, ↓ TCR αβ/↑ TCR γδ cells; ↑ CD21lo B cells; ↓ NK, MAIT cells; no NKT cells; ↓ EBV-specific CD8+ T cells | Rituximab, IV Ig, allogeneic HSCT (1 patient) | ↓ ERK activation in T cells; ↓ T cell proliferation to mitogen, Ags; ↓ CD27/CD70-induced T cell proliferation; ↓ CTPS1 expression in activated T cells; ↓ NK cell function | 61-65 |
| CD27 (n = 18) | Age of onset, 1-22 y EBV viremia (∼90%), severe HLH (∼25%), EBV-induced lymphoproliferation (∼50%), EBV+ B lymphoma (∼40%; Hodgkin, DLBCL), lymphadenopathy, hypogammaglobulinemia, poor Ab responses to some vaccines/infections (∼70%), autoimmunity (20-25%) | EBV,VZV, CMV; recurrent bacterial/viral infections | ∼30% mortality (5/18 patients deceased) | Normal/↓ NKT, MAIT cells; ↓ EBV-specific CD8+ T cells; ↓ 2B4 expression on CD8+ T cells | Rituximab, R-CHOP, steroids; IV Ig; 3/3 patients successfully treated with HSCT | ↓ CD27/CD70-induced T cell proliferation; ↓ EBV Ag-induced T cell proliferation/expansion; ↓ CD8+ T cell cytotoxicity toward B cells; ↓ NKG2D, 2B4 may impair CD8+ T cell cytotoxicity toward B cells | 66-69 |
| CD70 (n = 6) | Age of onset, 1-5 y EBV viremia (100%), EBV lymphoproliferation (80%), EBV+ B lymphoma (Hodgkin; 80%), lymphadenopathy, hypogammaglobulinemia, poor Ab responses to some vaccines/infections (60-80%), autoimmunity (20-25%, eg Behçet) | EBV,VZV, CMV (50%);recurrent bacterial/viral infections | All patients currently alive | ↑ naive CD4+, CD8+ T cells; ↓ memory B cells; normal/↓ EBV-specific CD8+ T cells but impaired functionality; ↓ NKG2D, 2B4 on CD8+ T cells | IV Ig, prophylactic antibiotics; chemotherapy for lymphoma; rituximab; 2 patients successfully treated with HSCT | 69-71 | |
| TNFRSF9* (n = 8) | Age of onset, 3-6 y EBV viremia (75%), HLH (25%), EBV lymphoproliferation (50%), EBV+ B lymphoma (3 patients), EBV T-cell lymphoproliferation (CAEBV), recurrent sinopulmonary infections (Candida, Staphylococcus, pneumonia; 90%), lymphadenopathy. hypogammaglobulinemia, poor Ab responses to some vaccines/infections (∼60%) | EBV, HSV | 7 patients currently alive, 1 died (HLH) | ↓ naive, ↑ effector memory CD4+, CD8+ T cells (post-chemo/rituximab); ↓ memory B cells; ↓ NK, NKT, MAIT cells | IV Ig; rituximab; chemotherapy for lymphoma; HSCT (1 patient) | ↓ T cell proliferation, IFN-γ secretion; ↓ EBV-specific CD8+ T-cell cytotoxicity | 72-74 |
| Mutated gene | Clinical features | Viral infections | Outcomes | Cellular phenotype | Treatment | Mechanism of disease | References |
|---|---|---|---|---|---|---|---|
| SH2D1A (SAP) (n > 100) | Age of onset, 0.5-40 y EBV viremia (75%), FIM/HLH (∼50-60%), hypogammaglobulinemia (40-50%), B lymphoma (Burkitt/NHL; EBV+ and EBV−; ∼25%) | EBV | ∼30-80% mortality | ↓ memory B cells, no NKT cells, ↑ TEMRA (exhausted-type) CD8+ T cells | IV Ig, B-cell depletion (rituximab), chemotherapy for lymphoma, HSCT (curative in >90% of cases) | ↓SLAM/SAP-dependent activation of NK and Ag-specific CD8+ T cells by Ag-presenting or transformed B cells | 26-34 |
| ITK (n = 22) | Age of onset, 3-44 y EBV viremia (100%), EBV-induced lymphoproliferation (∼70%), EBV+ B-cell lymphoma (mostly Hodgkin; 1 NHL, 1 Burkitt lymphoma) (∼80%), other infections (epidermodysplasia verruciformis; 2 patients), CD4+ T-cell lymphopenia (∼65%), splenomegaly (∼60%), progressive hypogammaglobulinemia (>50%), some autoimmunity (25%) | EBV,CMV, VZV,HPV | 45% mortality | Normal % CD3+ T cells but ↓ naive CD4+ T cells, ↓ NKT cells | Rituximab, chemotherapy for lymphoma, IV Ig (not particularly effective), HSCT (5/7 alive and well post-HSCT) | ↓T cell activation (↓PLCγ activation) | 35-47 |
| MAGT1 (n = 22) | Age of onset, 3-45 y, chronic EBV viremia (>90%), EBV-induced lymphoproliferation (not HLH), EBV+ B lymphoma (Burkitt/Hodgkin/DLBCL; 70%), CD4+ T-cell lymphopenia (∼50%), splenomegaly, progressive hypogammaglobulinemia (>50%), intellectual/developmental disability (2/3 patients), autoimmunity (cytopenia, Guillain-Barre syndrome, hepatitis), recurrent infections (80%), Kaposi sarcoma (1 patient), Castleman disease (1 patient) | EBV,VZV, HSV, CMV, KSHV; molluscum contagiosum | ∼25% mortality | ↑ B cells, ↓ memory B cells, inverted CD4/CD8 ratio, ↓ NKG2D expression on NK and CD8+ T cells | Rituximab, R-CHOP; IV Ig; HSCT (2/4 patients died post HSCT); risk of hemorrhage pre-/post-HSCT; Mg supplementation (though variable effect) | ↓T cell activation (↓ TCR-induced Mg2+/Ca2+ flux, PLCγ activation); ↓ NKG2D impairs NK/CD8+ T cells cytotoxicity toward B cells; glycosylation defect (likely impairs expression of NKG2D, and other important immunoregulatory glycoproteins, as well as potentially affecting stability of serum Ig) | 46-56 |
| CTPS1 (n = 18) | Age of onset: 0-5 y EBV viremia (> 90%) EBV-lymphoproliferative diseases (30-50%) Severe IM (∼50%) Recurrent viral/bacterial infections (100%) Poor Ab responses to vaccines/infections; hypogammaglobulinemia (∼30%) | EBV,VZV (∼50%), HSV, CMV, HHV6 (∼20%); norovirus, adenovirus | ∼40% mortality | ↓ NKT, NK, MAIT cells; ↓ memory B and CD8+ T cells | Rituximab, adoptive EBV-specific CD8+ T cell therapy, IV Ig, HSCT (11/14 alive and well) | ↓ activation/proliferation of T and B cells (↓ de novo pyrimidine synthesis), CD27/CD70-induced T cell proliferation may be impaired, ↓IFN-γ production | 57-60 |
| RASGRP1 (n = 9) | Age of onset, 0-7 y EBV viremia (>70%) EBV+ B-lymphoma (Hodgkin), smooth muscle tumor (∼70%) CD4+ T cell lymphopenia Poor Ab responses to some vaccines/infections (∼20%) Epidermodysplasia verruciformis (1 patient) Lymphadenopathy Autoimmunity (cytopenias, ANA, hepatitis; ∼50%) Recurrent bacterial/viral infections (pneumonia) (100%) HLH (1 patient) | EBV,CMV, VZV, HSV1,HPV;molluscum contagiosum | ∼30-35% mortality (3/9 patients deceased) | ↓ naive CD4+ and CD8+ T cells, ↓ TCR αβ/↑ TCR γδ cells; ↑ CD21lo B cells; ↓ NK, MAIT cells; no NKT cells; ↓ EBV-specific CD8+ T cells | Rituximab, IV Ig, allogeneic HSCT (1 patient) | ↓ ERK activation in T cells; ↓ T cell proliferation to mitogen, Ags; ↓ CD27/CD70-induced T cell proliferation; ↓ CTPS1 expression in activated T cells; ↓ NK cell function | 61-65 |
| CD27 (n = 18) | Age of onset, 1-22 y EBV viremia (∼90%), severe HLH (∼25%), EBV-induced lymphoproliferation (∼50%), EBV+ B lymphoma (∼40%; Hodgkin, DLBCL), lymphadenopathy, hypogammaglobulinemia, poor Ab responses to some vaccines/infections (∼70%), autoimmunity (20-25%) | EBV,VZV, CMV; recurrent bacterial/viral infections | ∼30% mortality (5/18 patients deceased) | Normal/↓ NKT, MAIT cells; ↓ EBV-specific CD8+ T cells; ↓ 2B4 expression on CD8+ T cells | Rituximab, R-CHOP, steroids; IV Ig; 3/3 patients successfully treated with HSCT | ↓ CD27/CD70-induced T cell proliferation; ↓ EBV Ag-induced T cell proliferation/expansion; ↓ CD8+ T cell cytotoxicity toward B cells; ↓ NKG2D, 2B4 may impair CD8+ T cell cytotoxicity toward B cells | 66-69 |
| CD70 (n = 6) | Age of onset, 1-5 y EBV viremia (100%), EBV lymphoproliferation (80%), EBV+ B lymphoma (Hodgkin; 80%), lymphadenopathy, hypogammaglobulinemia, poor Ab responses to some vaccines/infections (60-80%), autoimmunity (20-25%, eg Behçet) | EBV,VZV, CMV (50%);recurrent bacterial/viral infections | All patients currently alive | ↑ naive CD4+, CD8+ T cells; ↓ memory B cells; normal/↓ EBV-specific CD8+ T cells but impaired functionality; ↓ NKG2D, 2B4 on CD8+ T cells | IV Ig, prophylactic antibiotics; chemotherapy for lymphoma; rituximab; 2 patients successfully treated with HSCT | 69-71 | |
| TNFRSF9* (n = 8) | Age of onset, 3-6 y EBV viremia (75%), HLH (25%), EBV lymphoproliferation (50%), EBV+ B lymphoma (3 patients), EBV T-cell lymphoproliferation (CAEBV), recurrent sinopulmonary infections (Candida, Staphylococcus, pneumonia; 90%), lymphadenopathy. hypogammaglobulinemia, poor Ab responses to some vaccines/infections (∼60%) | EBV, HSV | 7 patients currently alive, 1 died (HLH) | ↓ naive, ↑ effector memory CD4+, CD8+ T cells (post-chemo/rituximab); ↓ memory B cells; ↓ NK, NKT, MAIT cells | IV Ig; rituximab; chemotherapy for lymphoma; HSCT (1 patient) | ↓ T cell proliferation, IFN-γ secretion; ↓ EBV-specific CD8+ T-cell cytotoxicity | 72-74 |
Ag, antigen; CAEBV, chronic active EBV; DLBCL: diffuse large B-cell lymphoma; HPV, human papillomavirus; Ig, immunoglobulin; KSHV, Kaposi sarcoma herpesvirus; MAIT, mucosal-associated invariant T; Mg, magnesium; NHL, non-Hodgkin lymphoma; R-CHOP, rituximab, cyclophosphamide, doxorubicin hydrochloride, vincristine sulfate, prednisone.
Eleven individuals with recessive mutations in TNFRSF9 have been identified, but 4 were asymptomatic. However, 1 of these unaffected individuals is included in the table, as the patient did have chronic EBV viremia.
XLP1 due to inactivating mutations in SH2D1A
X-linked lymphoproliferative disease (XLP) is the textbook example of a PID with a selective inability to generate effective immunity against EBV infection in otherwise healthy individuals. XLP1 was first reported in the 1970s by Bar et al,75 Purtillo et al,76 and Provisor et al77 in 13 males from 3 unrelated families presenting with an often-fatal immunodeficiency associated with EBV infection.75-77 Strikingly, these same patients exhibited intact immunity against other childhood viruses (HSV, VZV, and CMV).7-9,25,75-77 The defining features of XLP1 are severe EBV-induced IM/HLH (hepatosplenomegaly, extensive tissue damage, and organ failure), B lymphoma, and dysgammaglobulinemia, with the latter features being EBV independent26-29 (Table 1; Figure 1A-B). Age of onset is ∼5 years (Figure 1C),27,29 and hematopoietic stem cell transplant (HSCT) is curative, with 80% to 95% survival >5 years post-HSCT27,30 (Table 1; Figure 1D). The greatest mitigating risk for successful HSCT was HLH pretransplant. In HLH+ patients, survival post-HSCT was 50%, compared with >90% for XLP patients who had not developed HLH. Overall survival of untransplanted patients is 20% to 50%, underscoring the highly favorable outcomes of HSCT for XLP1 if a compatible donor is available.27,29,30 XLP1 has also been treated with B-cell depletion, which efficiently reduces EBV viremia by eliminating the viral host cell27,78 (Table 1). Rituximab was also used to treat B lymphoma in males subsequently found to have XLP179 and has been successfully used during conditioning for HSCT.27,80
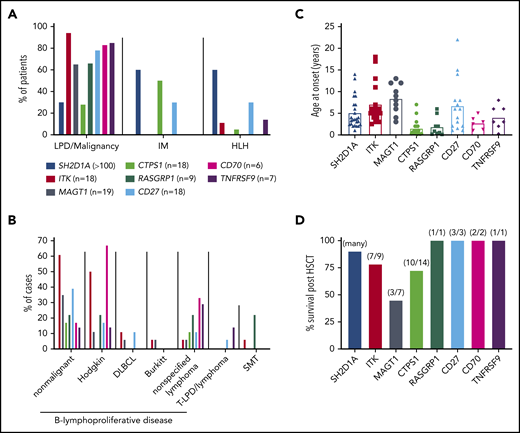
Clinical features of inborn errors of immunity predisposing to EBV-induced disease. (A) Proportions of patients with mutations in the indicated genes developing lymphoproliferative disease (LPD)/malignancy, severe IM, and/or HLH. (B) Breakdown of lymphoproliferative disease and the types of malignancies observed in these patients. DLBCL, diffuse large B cell lymphoma; SMT, smooth muscle tumor; T-LPD, T cell lymphoproliferative disease. Data for XLP1 is not included in this graph. (C) Age of onset of initial presenting clinical features for the indicated genotypes. Data for 25 individual patients with XLP1 (SH2D1A mutations) are derived from Pachlopnik Schmid et al.29 The average for these 25 patients (4.9 years) is consistent with the median reported by Booth et al27 for large cohorts of EBV+ (4 years; n = 51) and EBV− (3 years; n = 28) XLP1 patients. (D) Survival post-HSCT. Data for XLP1 patients is based on the study by Booth et al27 ; data for the other genotypes are from individual case reports. Note that apart from XLP1 due to SH2D1A mutations, relatively few patients with other monogenic mutations have undergone HSCT.
Clinical features of inborn errors of immunity predisposing to EBV-induced disease. (A) Proportions of patients with mutations in the indicated genes developing lymphoproliferative disease (LPD)/malignancy, severe IM, and/or HLH. (B) Breakdown of lymphoproliferative disease and the types of malignancies observed in these patients. DLBCL, diffuse large B cell lymphoma; SMT, smooth muscle tumor; T-LPD, T cell lymphoproliferative disease. Data for XLP1 is not included in this graph. (C) Age of onset of initial presenting clinical features for the indicated genotypes. Data for 25 individual patients with XLP1 (SH2D1A mutations) are derived from Pachlopnik Schmid et al.29 The average for these 25 patients (4.9 years) is consistent with the median reported by Booth et al27 for large cohorts of EBV+ (4 years; n = 51) and EBV− (3 years; n = 28) XLP1 patients. (D) Survival post-HSCT. Data for XLP1 patients is based on the study by Booth et al27 ; data for the other genotypes are from individual case reports. Note that apart from XLP1 due to SH2D1A mutations, relatively few patients with other monogenic mutations have undergone HSCT.
XLP1 is caused by mutations in SH2D1A, encoding the SH2-domain containing cytoplasmic adaptor protein SAP (SLAM-associating protein).81-83 SAP links SLAM-family receptors on hematopoietic cells with downstream intracellular signaling pathways to regulate effector functions of T and NK cells and NKT cell development.84-86 In the absence of SAP, cytotoxic functions of CD8+ T and NK cells induced by engaging the SLAM-family receptors 2B4 (CD244) and NTB-A are abolished31-33,87-89 (Figure 2). Furthermore, NKT cells are absent from XLP1 patients and Sh2d1a gene–targeted mice90,91 (Table 1). The defect in responses of SAP-deficient CD8+ T and NK cells following engagement of SLAM receptors, but not other activating receptors, mirrors XLP patients being extremely susceptible to EBV-induced disease but having intact immunity against other pathogens (Table 1). Similarly, when antiviral immune responses were tracked in female carriers of XLP1, CMV- and influenza-specific CD8+ T cells were detected in SAP− and SAP+ subsets of memory CD8+ T cells, yet EBV-specific CD8+ T cells were mostly (>95%) SAP+.30,33 These dichotomous responses raised the question of how SAP deficiency uniquely renders affected individuals exquisitely vulnerable to EBV infection, which typically runs a benign course in most healthy individuals.
![Molecular and biochemical requirements for inducing CD8+ T cell-mediated EBV-specific immunity. The scheme outlines the key cell surface receptor–induced signaling pathways required for generating CD8+ T-cell–mediated immunity against EBV, and how specific inborn errors of immunity (SH12D1A [SAP], MAGT1, ITK, CD27, CD70, TNFRSF9 [4-1BB], RASGRP1, and CTPS1; depicted in red) can compromise these processes. (A) Initial interactions between EBV-infected or antigen-presenting B cells and CD8+ T cells involve CD27/CD70, 4-1BB/4-1BBL, and major histocompatibility complex (MHC) class I/TCR. Signals elicited downstream of these receptors, requiring MAGT1, ITK, and RasGRP1, induce DNA synthesis via induction and activation of CTPS1 and subsequent proliferation of EBV-specific CD8+ T cells. Signals via CD27/CD70 and MAGT1 contribute to inducing or maintaining expression of activating receptors such as 2B4 and NKG2D. (B) Following expansion of EBV-specific CD8+ T cells, engagement of the activating SLAM family receptors 2B4 and NTB-A (via SAP) and the glycoprotein NKG2D by their ligands highly expressed on EBV-infected B cells induces cytolytic effector function, resulting in CD8+ T-cell–mediated killing of EBV+ target B cells.](https://ash.silverchair-cdn.com/ash/content_public/journal/blood/135/9/10.1182_blood.2019000928/4/m_bloodbld2019000928cf2.png?Expires=1769103160&Signature=GnGL5nMi5uutNuhhvS1Aq7tXxKK4TI9Hd384M6cKFlM1ypO1xHXUe06R3Bz0XJC5AOBpLzSUaZ0RhKlc4bqchvtP6heNAr1mWlXXouDu54XAyWsC25oWsJnbpLpPSVB0K18dqhMMjHflWmgj1W8hjDmSEM3iciS8SwsJjfcFhDFU0k4hjcbLxqTLevcKid5owNf8tMmu3J~1sgheJl50YlQ9NMAySiUBZTH3L1hEk3a6uG7H1bRqTbQLwoR0zjQdltiZVJ02b9tvLK9zEgNhwOH3~ebnNUfzgsUuaakPzZ4kjZkXqVtfSbDWRXe574rUH5nGU6NnoTxh2awvAB~nug__&Key-Pair-Id=APKAIE5G5CRDK6RD3PGA)
Molecular and biochemical requirements for inducing CD8+ T cell-mediated EBV-specific immunity. The scheme outlines the key cell surface receptor–induced signaling pathways required for generating CD8+ T-cell–mediated immunity against EBV, and how specific inborn errors of immunity (SH12D1A [SAP], MAGT1, ITK, CD27, CD70, TNFRSF9 [4-1BB], RASGRP1, and CTPS1; depicted in red) can compromise these processes. (A) Initial interactions between EBV-infected or antigen-presenting B cells and CD8+ T cells involve CD27/CD70, 4-1BB/4-1BBL, and major histocompatibility complex (MHC) class I/TCR. Signals elicited downstream of these receptors, requiring MAGT1, ITK, and RasGRP1, induce DNA synthesis via induction and activation of CTPS1 and subsequent proliferation of EBV-specific CD8+ T cells. Signals via CD27/CD70 and MAGT1 contribute to inducing or maintaining expression of activating receptors such as 2B4 and NKG2D. (B) Following expansion of EBV-specific CD8+ T cells, engagement of the activating SLAM family receptors 2B4 and NTB-A (via SAP) and the glycoprotein NKG2D by their ligands highly expressed on EBV-infected B cells induces cytolytic effector function, resulting in CD8+ T-cell–mediated killing of EBV+ target B cells.
Molecular and biochemical requirements for inducing CD8+ T cell-mediated EBV-specific immunity. The scheme outlines the key cell surface receptor–induced signaling pathways required for generating CD8+ T-cell–mediated immunity against EBV, and how specific inborn errors of immunity (SH12D1A [SAP], MAGT1, ITK, CD27, CD70, TNFRSF9 [4-1BB], RASGRP1, and CTPS1; depicted in red) can compromise these processes. (A) Initial interactions between EBV-infected or antigen-presenting B cells and CD8+ T cells involve CD27/CD70, 4-1BB/4-1BBL, and major histocompatibility complex (MHC) class I/TCR. Signals elicited downstream of these receptors, requiring MAGT1, ITK, and RasGRP1, induce DNA synthesis via induction and activation of CTPS1 and subsequent proliferation of EBV-specific CD8+ T cells. Signals via CD27/CD70 and MAGT1 contribute to inducing or maintaining expression of activating receptors such as 2B4 and NKG2D. (B) Following expansion of EBV-specific CD8+ T cells, engagement of the activating SLAM family receptors 2B4 and NTB-A (via SAP) and the glycoprotein NKG2D by their ligands highly expressed on EBV-infected B cells induces cytolytic effector function, resulting in CD8+ T-cell–mediated killing of EBV+ target B cells.
This paradox was resolved by analyzing responses of SAP− and SAP+ CD8+ T cells specific for the same antigens presented on different antigen-presenting cells (APCs). Thus, while killing of dendritic cells (DCs), monocytes, or fibroblasts was unaffected by SAP deficiency, the ability of CD8+ T cells to respond to B cells required SAP signaling downstream of 2B4 and NTB-A.32,33 (Figure 2B) Similar findings were observed for CD8+ T cells from Sap-deficient mice, which killed Ag-presenting fibrosarcoma, but not B cells.92 This resulted from defective immunological synapse formation between Sap-deficient CD8+ T cells and B-cell targets and reduced activation of Src kinases in CD8+ T cells, diminishing cytotoxicity specifically toward B cells.92 Thus, these SLAM receptors have critical and nonredundant roles in SAP-mediated co-stimulation of Ag-specific CD8+ T cells. Consistent with this, ligands of these receptors are highly expressed on B cells, but not fibroblasts or DCs.32,33
The importance of SLAM/SAP signaling in CD8+ T cells for immunity against EBV elicited by Ag-presenting B cells was further, and serendipitously, revealed by the discovery of somatic reversion and subsequent gene repair in XLP1 patients.30,34 Here, the pathogenic mutation in SH2D1A was corrected in some lymphoid cells (predominantly memory CD8+ T cells), allowing detectable expression of SAP. While both SAP-deficient and reverted SAP+ CD8+ T cells from these XLP1 patients responded to polyclonal stimuli, responses to EBV-presenting B cells were almost exclusively derived from the minor population of revertant SAP+ CD8+ T cells.30,34
These findings revealed that the inability of XLP1 patients to control EBV infection results from crippled interactions between SAP-deficient cytotoxic cells and Ag-presenting or virally infected B cells and subsequent impaired B-cell mediated activation of these cells (Figure 2B). This results from the unique B-cell tropism of EBV, whereas many other viruses infect >1 host cell type.1,4,6 Consequently, in the setting of infection with viruses other than EBV, alternate APC-driven activation pathways exist, enabling the host to generate protective immune responses even in the presence of SH2D1A mutations. Thus, SLAM/SAP signaling is critical for protective immunity against EBV but is redundant for immunity against other infections.
Genetic lesions in regulators of TCR signaling predispose to EBV-induced disease
Engagement of the T cell receptor (TCR) initiates a complex series of biochemical events that activate numerous signaling pathways, ultimately resulting in the expansion and differentiation of Ag-specific T cells.93 Inborn errors in distinct but interacting components of TCR-induced signaling pathways underlie predisposition to severe EBV-induced disease in specific PIDs. This highlights the fundamental requirement for extensive T-cell expansion following acute EBV exposure to efficiently protect the host from the deleterious effects of viral infection.
ITK and MAGT1 deficiency
ITK encodes IL-2–inducible T-cell kinase, a nonreceptor kinase involved in proximal TCR signaling.93 ITK coordinates T-cell activation by regulating PLCγ1 phosphorylation and subsequent calcium release.93 MAGT1 encodes a ubiquitously expressed magnesium transporter.48 By moderating intracellular Mg2+ levels, MAGT1 acts as a second messenger in TCR-mediated activation.48 Loss-of-function mutations in ITK and MAGT1 were first reported in 200935 and 2011,49 respectively. To date, 22 patients with recessive ITK35-45 or hemizygous MAGT1 mutations48-56 (S.L., unpublished data) have been described in cohort studies, case reports, or outcomes from next-generation sequencing efforts in different centers46,47 (Table 1). There is strong overlap in the clinical phenotype and age of onset of ITK- and MAGT1-deficient patients. Thus, EBV viremia, EBV-induced lymphoproliferative disease, and/or EBV+ B lymphoma (Hodgkin, non-Hodgkin [including Burkitt and diffuse large B cell lymphoma]) have been reported for all but 1 ITK-deficient patient35-45 and 19 out of 22 MAGT1-deficient patients,48-56 with B-cell malignancies occurring in 70% to 80% of all patients (Table 1; Figure 1A-C).
Additional clinical features of these PIDs are CD4+ T lymphopenia and hypogammaglobulinemia. Unlike XLP, patients with ITK or MAGT1 mutations are prone to infections with other viral (eg, VZV, CMV, HSV, KSHV, β-HPV, and Molluscum contagiosum) and bacterial (Pneumococcus and Haemophilus) pathogens35-45,48-56,80 (Table 1). However, the major pathogenic threat to these patients is unquestionably EBV, even though HLH is less frequent than XLP1 (Figure 1A). Interestingly, 1 ITK-deficient patient was identified at age 6 years with CD4+ lymphopenia and hypogammaglobulinemia but was EBV negative until their late teens.39 We also identified an EBV− MAGT1-deficient patient presenting with HHV8− Castleman disease, CD4+ lymphopenia, and hypogammaglobulinemia (Klinken et al94 ; Table 1). Without major intervention, ITK or MAGT1 deficiency can be fatal. Indeed, 8 out of 22 ITK-deficient patients died prior to HSCT. Of the 9 who underwent HSCT, 7 are currently alive (overall survival, ∼55%; Figure 1D).35-45,80 Survival for MAGT1-deficiency is better, as only 1 untransplanted patient died.51 However, 4 out of 7 patients did not survive post-HSCT (overall survival, ∼75%; Figure 1D; Table 1).48,54,56
Magnesium supplementation was used to treat 2 XMEN patients, resulting in fewer EBV-infected B cells.48,50 However, we found minimal clinical improvement in 3 MAGT1-deficient patients following oral magnesium administration (Klinken et al94 ; S.L., unpublished data). To determine the efficacy of magnesium supplementation, a larger number of patients will need to be enrolled in a formal clinical trial and the subsequent findings provided to the research and clinical communities.
Despite the similar clinical features, survival is better for MAGT1 than ITK deficiency. This presumably reflects prominent functions of ITK in fundamental aspects of lymphocyte development that are not shared by MAGT1. ITK plays a role in T-cell development and selection and is required for cytoskeletal rearrangements and cell polarization for the release of cytokines and cytolytic mediators.93 Interestingly, survival post-HSCT is superior for ITK deficiency compared with MAGT1 deficiency (Figure 1D); this may reflect hemorrhagic complications due to MAGT1 mutations.54
MAGT1 mutations were recently identified in 2 unrelated individuals who presented with intellectual and developmental disability associated with defective N-linked glycosylation but no signs of immunodeficiency.55 This study also reported glycosylation defects in a third patient who had typical XMEN immunodeficiency but no evidence of intellectual or developmental impairment.55 This led the authors to propose MAGT1 deficiency is a novel congenital disorder of glycosylation (CDG).55 However, it is unknown how variants in the same gene yield such clinically diverse phenotypes. This divergence was not explained by the nature of the variant, as distinct loss-of-expression mutations resulted in immunodeficiency in 1 patient but CDG in another.55 Identification of additional MAGT1 patients, with or without intellectual/developmental disability, will be required to further this hypothesis.
Analysis of functional defects in immune cells from ITK- or MAGT1-deficient individuals identified critical roles for these molecules in T-cell signaling and revealed mechanisms of disease pathogenesis due to these variants. ITK or MAGT1 mutations generally diminished TCR-induced Ca2+ flux and subsequent PLCγ1 activation, compromising T-cell activation, proliferation, and function36,39,44,49,53 (Table 1; Figure 2). A surprising finding was reduced expression of the C-type lectin NKG2D on MAGT1-deficient NK and CD8+ T cells50 (Figure 2A). This phenotype can identify individuals with MAGT1 mutations, confirm the pathogenic effects of novel MAGT1 variants, and track donor lymphocyte reconstitution in patients following HSCT.51-55,94 As NKG2D is heavily glycosylated,95 reduced expression on MAGT1-deficient cells probably results from MAGT1 deficiency representing a CDG.55
Reduced NKG2D expression was sufficient to compromise cytotoxicity of MAGT1-deficient CD8+ T and NK cells against EBV+ B cells,50 which express high levels of NKG2D ligands95 (Figure 2B). Thus, similar to XLP1 and SLAM receptors,7,28 MAGT1 deficiency confirmed the critical role of NKG2D signaling in inducing cytolytic function of CD8+ T and NK cells (Table 1). The similar clinical, cellular, biochemical, and functional phenotypes arising from MAGT1 and ITK mutations suggest ITK-deficient cells will also exhibit impaired killing of EBV-B cells due to poor proliferation and cognate interactions resulting from disrupted function and/or expression of receptors such as 2B4, NTB-A, or NKG2D.
CTPS1 deficiency
CTPS1 encodes cytidine 5′ triphosphate (CTP) synthase 1 (CTPS1), an enzyme required for synthesizing CTP, a key component of nucleic acids. As such, CTPS1 is required for DNA replication.8 Recessive mutations in CTPS1 predispose to severe EBV infection and associated lymphoproliferative disorders57 (Figure 1A-B). CTPS1 deficiency due to a unique homozygous mutation was originally identified in 8 patients, resulting from a founder effect in the population of northwest England.57 Additional patients (n = 18) have since been reported with the same founder mutation and similar clinical presentations58-60 (S.L., unpublished data; Table 1). Similar to ITK and MAGT1 deficiency, individuals with CTPS1 mutations experience recurrent severe infections with non-EBV pathogens (VZV, HHV6, and bacteria).57-60 However, age of onset is generally earlier in individuals with CTPS1 mutations compared with those with SH2D1A, ITK, or MAGT1 mutations (Figure 1C). Two CTPS1-deficient patients died of EBV-related disease.57 While 14 patients successfully received HSCT, 4 died of complications or viral infections following HSCT (Figure 1D). Two patients are alive without HSCT.57-60
The CTPS1 mutation causes protein instability without affecting enzymatic activity.8,57 Thus, patients have 80% less CTPS1 catalytic activity than healthy individuals (S.L., unpublished data). CTPS1 deficiency dramatically decreases TCR-induced proliferation, yet effector functions are preserved8,57 (Table 1; Figure 2A). CTPS1 expression is low in resting T cells but is rapidly induced following TCR activation, consistent with its requirement for DNA synthesis and proliferation.8,57 This highlights the selective role of CTPS1 in T-cell proliferation, which is required to sustain massive T-cell expansion following EBV infection in healthy individuals4,6,16,17,19 (Figure 2A).
RASGRP1 deficiency
RAS guanyl-releasing protein 1 (RASGRP1) is a guanine nucleotide exchange factor highly expressed in NK and T cells that is selective for the small GTPase protein Ras.96 RASGRP1 is considered the main activator of the Ras/MAPK pathway in T and NK cells.96 Nine patients have been reported with biallelic RASGRP1 loss-of-function mutations and a combined immunodeficiency characterized by susceptibility to EBV, CMV, HSV1, HPV, and some fungal and/or bacterial pathogens.61-65 However, all cases presented with severe EBV-driven lymphoproliferative disorders, including B lymphoma (n = 6), and autoimmune manifestations61-65 (n = 5; Table 1; Figure 1A-B). Two patients died of malignancy and 1 of thrombocytopenic purpura. Three patients underwent HSCT, including 2 autologous HSCTs leading to remission of lymphoma (Table 1). The impact of autologous HSCT on the consequences for EBV-induced disease is unknown61-65 (Figure 1D).
RASGRP1-deficient T cells exhibited decreased MAPK activation and impaired proliferation (Figure 2A). EBV-specific T cells were reduced in 1 patient, suggesting limited expansion of these cells64 (Table 1). RASGRP1-deficient T and NK cells were originally found to have decreased killing due to impaired cytotoxic granule exocytosis.61 However, another study found intact degranulation of RASGRP1-deficient CD8+ T cells.64 RASGRP1-deficient T cells also have diminished migratory capacity associated with defective activation of RhoA.61 TCR excision circles and TCR-β repertoire diversity were reduced and TCR-γδ frequencies increased in 2 patients, suggesting impaired TCR rearrangement during thymic maturation may account for autoimmune manifestations.65 Rasgrp1-deficient mice also develop autoimmunity, possibly due to abnormal pre-TCR signaling and positive selection, which are RASGRP1 dependent.8
Susceptibility of RASGRP1-deficient individuals to EBV-driven lymphoproliferation and malignancy likely results from defective expansion and function of EBV-specific T cells (Figure 2A). Interestingly, RASGRP1-deficient T cells have reduced CTPS1 expression,64 while 2 patients with ITK mutation43 and 1 with RASGRP1 mutation63 developed epidermodysplasia verruciformis due to HPV infection, in addition to EBV+ B lymphoma. These and other similarities in patients with mutations affecting proximal (ITK and MAGT1) or distal (RASGRP1) TCR signaling underscore the critical role of T-cell expansion in controlling EBV infection (Figure 2A).
TNF receptor (TNFR)/TNF superfamily members have critical roles in initiating EBV-specific CD8+ T cell responses
The study of XLP1 and MAGT1 deficiency elucidated fundamental requirements for interactions between activating receptors on cytotoxic cells and corresponding ligands on cognate B cells in controlling EBV infection.7,28,48 Further analyses of patients with EBV-associated PIDs have identified additional receptor/ligand pairs with nonredundant functions in cell-mediated anti-EBV immunity.
CD27/CD70 deficiency
Ten patients from four kindreds who developed combined immunodeficiency characterized by EBV viremia, lymphoproliferation, HLH, EBV+ B lymphoma, hypogammaglobulinemia, and impaired Ab responses were reported in 2012 and 2013.66,67 These individuals had homozygous mutations in CD27,66,67 a TNFR superfamily (TNFRSF) member expressed by many immune cells, including T, B, and NK cell subsets.7,97 Since these reports, 7 additional patients from 5 unrelated families have been reported with biallelic (6 homozygous and 1 compound heterozygous) CD27 mutations66-69 (Figure 1A-B). One patient with a heterozygous mutation was also identified68 (Table 1). As heterozygous parents and siblings of patients with homozygous CD27 mutations are unaffected, this patient probably has a cryptic variant on the other CD27 allele. Indeed, CD27 was undetectable on their lymphocytes, while it was present on cells from the bona fide heterozygous father.68 EBV-induced disease was evident in >90% (16/18) of patients and fatal in 25% to 30% of cases (Figure 1A-B). B-cell depletion was effective at reducing EBV loads and improving symptoms in 4 out of 4 patients, and 3 patients successfully underwent HSCT (Figure 1D).67,68 Several CD27-deficient patients also suffered recurrent bacterial and herpesvirus (CMV and VZV) infections66-69 (Table 1).
CD27 binds CD70, a TNFSF ligand predominantly expressed on activated B cells, myeloid cells, some T cells,97 and EBV-infected B cells in IM patients.69 Since 2017, homozygous CD70 mutations that affect expression or binding to CD27 have been identified in 6 individuals from 4 families.69-71 These patients had severe EBV viremia, with most developing EBV-associated lymphoproliferation (5/6), EBV+ B-lymphoma (Hodgkin [4/6]), and hypogammaglobulinemia (4/5) (Figure 1A-B); VZV infection occurred in 1 patient (Table 1). EBV viremia in CD70 deficiency was managed by rituximab in 3 patients,69-71 while 2 successfully underwent HSCT (Figure 1D), with CD70 expression on donor-engrafted cells being demonstrated for 1 patient69,71 (Table 1). Interestingly, somatic CD70 mutations have been detected in different B lymphomas,94,98,99 further linking a critical role for CD27/CD70 interactions in tumor immunity.
Mechanistically, CD8+ T cells from CD70-deficient patients were unable to efficiently lyse autologous EBV-infected B cells.69,70 CD27/CD70 interactions were critical for initial priming and expansion of EBV-specific CD8+ T cells rather than cytotoxicity69 (Figure 2A). This potentially explains reduced proportions of EBV-specific CD8+ T cells in some patients with CD7070 or CD27 mutations (S.L., unpublished data). Consistent with CD27 functioning as a co-stimulatory receptor on T cells and high CD70 expression on activated B cells,97 CD70 on B cells was required to engage CD27 on cognate CD8+ T cells to elicit a response69 (Figure 2A). Remarkably, CD70-deficient memory and EBV-specific CD8+ T cells have reduced expression of NKG2D and 2B470 ; we have made similar observations for CD27-deficient CD8+ T cells (S.L., unpublished data; Table 1, Figure 2). Thus, in addition to the inability of CD27- or CD70-deficient CD8+ T cells to be primed directly by CD27/CD70 interactions to proliferate, reduced expression of, and thus activation by, NKG2D and 2B4 could further compromise effector function of these cells toward EBV-infected B cells, akin to that observed for MAGT1- and SAP-deficient CD8+ T cells.7,28,48
Extending the observations of common pathways being impacted by distinct gene defects, RASGRP1-deficient T cells failed to proliferate in response to CD3/CD27 stimulation, suggesting Ras/MAPK is critical for not only CTPS1 expression57 but also CD27 signaling.95 These findings, together with comparably reduced proliferation of T cells with mutations in RASGRP162-65 or ITK,39,44 mechanistically link similarities in disease phenotype and pathogenesis due to mutations in CTPS1, RASGRP1, ITK, or CD27 (Figure 2A). MAGT1 deficiency may also compromise interactions between receptor-ligand pairs involved in anti-EBV-mediated immunity. While we did not observe reduced expression of CD27 on memory CD8+ T cells from MAGT1-deficient patients, we did report lower expression of CD28 on MAGT1-deficient CD8+ T cells.94 Furthermore, it was recently reported that CD70 expression on activated T cells is reduced under conditions of limiting magnesium, a mimic of MAGT1 defects.100 Thus, the effector function of MAGT1-deficient cytotoxic lymphocytes may be impaired not only by reduced NKG2D expression but also by insufficient engagement of CD27 and CD28 by their ligands CD70 and CD80/CD86 respectively, thereby resembling the phenotype of CD70-deficient T cells (Figure 2).
TNFRSF9/4-1BB/CD137 deficiency
Homozygous null mutations in another TNFRSF member, TNFRSF9 (4-1BB/CD137),97 have recently been identified in 8 individuals from 7 consanguineous kindreds. These patients presented with chronic EBV viremia, lymphoproliferation, lymphadenopathy, lymphoma, and HLH72-74 (Figure 1A-B). Recurrent bacterial infections and dysgammaglobuliemia were common in most affected individuals (Table 1).72-74 Interestingly, 2 siblings had persistent EBV-infected T cells associated with very high and chronic viremia, with 1 developing recurrent T lymphoproliferation and fatal HLH74 (Table 1; Figure 1A-B). Patients were treated with Ab replacement, antibiotics, and rituximab, while 1 patient successfully underwent HSCT72-74 (Figure 1D). Not surprisingly, viremia was not improved by rituximab in the individuals with EBV-infected T cells.74 Age of clinical onset in patients with TNFRSF9 mutations was comparable to the other PIDs reviewed here (Figure 1C).
CD137 has similar functions to CD27, enhancing T-cell proliferation, cytokine secretion, survival, and cytotoxicity.97 Unlike CD27, CD137 is not expressed by resting T cells; rather, it is induced following activation.97 CD137 ligand (CD137L/4-1BBL) is expressed by monocytes, DCs, and activated T and B cells.97 CD137-deficient patients exhibited impaired expansion of EBV-specific CD8+ T cells toward EBV-infected B cells (Figure 2A), reduced production of IFN-γ and perforin, and decreased mitochondrial activity and biogenesis72-74 (Table 1).
CD137-dependent proliferation was abrogated in CD137-deficient T cells but could be restored by ectopic expression of CD137.72,74 These observations suggest CD137 and CD27 have similar but nonredundant roles in EBV immunity and that the requirements to control EBV-infection in B and T cells overlap. It will be interesting to determine whether expression and/or function of 2B4 and NKG2D are affected by TNFRSF9 deficiency and whether mutations in TNFSF9 (CD137L) will be identified as another cause of EBV-susceptible PIDs. CD137 can also promote human NK cell proliferation.101 Given the possible contribution of NK cells to anti-EBV immunity,10-14 coupled with defects in NK cell function in PID patients susceptible to EBV-induced disease,7,50,88 impaired NK function may also contribute to disease due to CD137 mutations. Somatic TNFSF9 mutations have been found in some human B lymphomas,99 while CD137L-deficient mice exhibit increased incidence of B lymphoma compared with wild-type mice.102 Thus, mechanisms predisposing to hematological malignancies due to germline or somatic mutations in human CD137 could be further explored in CD137L-deficient mice.
In contrast to the other inborn errors discussed here, recessive TNFRSF9 mutations have incomplete clinical penetrance. Rodriguez et al reported that while both siblings in their study had chronically high EBV loads, 1 developed severe disease and fatal HLH while the other is clinically asymptomatic.74 Similarly, extended genotyping of family members of the 4 kindreds reported by Somekh et al identified 3 asymptomatic individuals with biallelic TNFRSF9 mutations.73 A possible explanation for incomplete penetrance was provided by Rodriguez et al, who discovered recessive inactivating mutations in PIK3CD, encoding the p110δ catalytic subunit of phosphatidylinositol 3-kinase, in addition to TNFRSF9, in the affected sibling but not the asymptomatic sibling with chronic EBV viremia.74 Thus, the severity of the clinical manifestations due to TNFRSF9 deficiency is likely influenced by additional genetic variants, such as polymorphisms or inborn errors of immunity. As defective proliferation of CD137-deficient T cells could be overcome by engaging CD28, OX40, or CD27,72,74 incomplete penetrance could result from differences in availability of co-stimulatory signals for activation of Ag-specific CD137-deficient T cells.
Conclusions
Although individually rare, in-depth analyses of PID patients have provided unprecedented advances in understanding molecular medicine, immune function, and development of therapies for treating diverse immune pathologies. PIDs have also revealed mechanisms underlying infectious disease and immune dysregulation and unequivocally identified nonredundant cellular, molecular, and biochemical pathways for host defense against specific pathogens and immune homeostasis. This has been clearly exemplified by studying patients with extreme susceptibility to EBV-induced pathology. While distinct genetic etiologies cause this phenotype, mechanisms of disease appear to intersect at several common points: (1) impaired activation/proliferation of EBV-specific CD8+ T cells (MAGT1, ITK, RASGRP1, CTPS1, CD27/CD70, and TNSRSF9) and (2) compromised provision of signals to induce cytotoxicity of CD8+ T and NK cells against EBV-infected B cells (SH2D1A, MAGT1, CD27/CD70, TNSRSF9, and RASGRP1) (Figure 2). Several of these genetic lesions impact both of these stages of eliciting cytotoxic immune responses. Furthermore, inputs for one pathway are required for the functioning of another (eg, RASGRP1-dependent induction of CTPS1; RASGRP1-dependent CD27-induced T cell proliferation; impaired NKG2D and 2B4 expression secondary to CD27/CD70 deficiency; and impaired PLCγ1 activation due to ITK or MAGT1 deficiency; Figure 2). These interconnections between different signaling modalities likely explain why one defined pathway is unable to compensate for impaired EBV-specific cytotoxic cell function arising from gene defects compromising another pathway.
There is an unusually high incidence of Hodgkin lymphoma in patients with ITK, RASGRP1, MAGT1, CD27/CD70, or CD137 mutations compared with XLP1 (Table 1; Figure 1B). While the reasons for this are unclear, these findings infer that SAP signaling is important for protection against multiple B-cell malignancies, yet CD137/CD137L, CD70-CD27/RASGRP1, or ITK/MAGT1 signaling is pivotal in preventing Hodgkin lymphoma. Lastly, the study of EBV-associated PIDs has revealed that the molecular requirements for interactions between CD8+ T cells and APCs differ substantially depending on the nature of the pathogen and the APC itself. Thus, the very nature of T-cell co-stimulation (ie, “signal 2”) is highly context dependent and cannot be generalized to invoke a predominant role by a single co-stimulatory receptor-ligand pair. Because of the oncogenic nature of EBV, elucidating mechanisms underlying recognition of transformed B cells in the context of EBV infection also have broad implications for understanding tumor surveillance and B-cell malignancies that arise independently of EBV. This is highlighted by the finding of somatic mutations in CD70 and TNFSF9 in EBV+ and EBV− human B-cell lymphomas.98,99 With the current rate of rapid discoveries of novel inborn errors of immunity, PIDs will continue to serve as unrivalled models linking genotype, clinical phenotype, and immune function and will allow the discovery of key tenets, and disprove some dogma, of innate and adaptive human immunity well into the future.
Acknowledgments
The authors are indebted to the patients and families who have made our research in this field possible, as well as all of their clinical and scientific colleagues with whom they have collaborated on this work over the years. S.G.T. thanks Evelyn Tangye for assistance in generating the figures.
The S.G.T. laboratory and Clincial Immunogenomics Research Consortium Australasia are supported by grants awarded by the National Health and Medical Research Council of Australia, the Cancer Council NSW, the Jeffrey Modell Foundation, the Ross Trust, the John Brown Cook Foundation, and the UNSW Sydney. The S.L. laboratory is supported by INSERM (France), Centre National de la Recherche Scientifique (France), Labélisation de la Ligue Nationale contre le Cancer (France), l’Agence Nationale de la Recherche (France), and the Société Française de Lutte contre les Cancers et Leucémies de l'Enfant et l'Adolescent.
Authorship
Contribution: S.G.T. and S.L. conceived and wrote this review.
Conflict-of-interest disclosure: The authors declare no competing financial interests.
Correspondence: Stuart G. Tangye, Garvan Institute of Medical Research, 384 Victoria St, Darlinghurst, Sydney 2010, NSW, Australia; e-mail: s.tangye@garvan.org.au.
