


Abstract
Our understanding of the genetics of acute myeloid leukemia (AML) development from myelodysplastic syndrome (MDS) has advanced significantly as a result of next-generation sequencing technology. Although differences in cell biology and maturation exist between MDS and AML secondary to MDS, these 2 diseases are genetically related. MDS and secondary AML cells harbor mutations in many of the same genes and functional categories, including chromatin modification, DNA methylation, RNA splicing, cohesin complex, transcription factors, cell signaling, and DNA damage, confirming that they are a disease continuum. Differences in the frequency of mutated genes in MDS and secondary AML indicate that the order of mutation acquisition is not random during progression. In almost every case, disease progression is associated with clonal evolution, typically defined by the expansion or emergence of a subclone with a unique set of mutations. Monitoring tumor burden and clonal evolution using sequencing provides advantages over using the blast count, which underestimates tumor burden, and could allow for early detection of disease progression prior to clinical deterioration. In this review, we outline advances in the study of MDS to secondary AML progression, with a focus on the genetics of progression, and discuss the advantages of incorporating molecular genetic data in the diagnosis, classification, and monitoring of MDS to secondary AML progression. Because sequencing is becoming routine in the clinic, ongoing research is needed to define the optimal assay to use in different clinical situations and how the data can be used to improve outcomes for patients with MDS and secondary AML.
Introduction
Myelodysplastic syndromes (MDSs) are a heterogenous group of clonal bone marrow disorders characterized by cytopenia, bone marrow dysplasia, ineffective hematopoiesis, and a high risk for transformation to acute myeloid leukemia (AML).1,2 MDS is typically diagnosed in older patients (median age, 70 years),3 and it is estimated that ≥45 000 new cases are diagnosed per year in the United States,4-7 making it one of the most common adult myeloid malignancies. By definition, MDS patients have a myeloblast count < 20%.
Approximately 30% of MDS patients eventually progress to AML, which is diagnosed by an increase in blast count to ≥20% of total nucleated cells in the bone marrow and is commonly termed “secondary AML to MDS.” Secondary AML accounts for up to 25% to 35% of total AML cases,8,9 with most (60-80%) arising from an antecedent MDS.10 The 2016 World Health Organization classification of hematologic malignancies classifies AML developing from MDS as a distinct clinicopathologic entity termed “AML with myelodysplasia-related changes” (AML-MRC).3 MDS patients who progress to secondary AML typically have inferior rates of complete remission, relapse-free survival, and overall survival compared with patients with de novo AML.8,9,11,12
Although MDS and secondary AML are classified as distinct entities, they represent a disease continuum that undergoes genetic clonal evolution. The discovery that similar genes are mutated in age-related clonal hematopoiesis (ARCH), also known as clonal hematopoiesis of indeterminate potential (CHIP), and myeloid malignancies suggests that progression from clonal hematopoiesis to MDS or AML is possible.13-22 Studies have shown that mutant cells in this spectrum of clonal myeloid diseases acquire additional mutations and clonally evolve during progression.13-15,17-21,23-27 Because of space constraints, this review will primarily focus on the genetic progression from MDS to secondary AML and address how monitoring for disease response and progression using sequencing could be implemented in the clinic.
Clinical and biologic aspects of MDS and secondary AML
MDS patients generally have a poor prognosis, with a median overall survival of only 5 years.28,29 This poor prognosis is not solely due to progression to secondary AML, indicating that MDS without progression is associated with mortality. In fact, as defined by the revised International Prognostic Scoring System,29 the overall survival of high-risk and very high-risk MDS (1.6 and 0.8 years, respectively)29 is similar to AML-MRC (10 months),12,30 indicating that higher-risk MDS and secondary AML are clinically similar. Making it clear that MDS and secondary AML represent cancers on a disease continuum can have a significant impact on a patient’s understanding of their disease.31,32
Cellular differences between MDS and secondary AML
Hematopoietic cell differentiation and maturation are abnormal in MDS and secondary AML. AML is characterized by a block in hematopoietic cell maturation, leading to an accumulation of myeloblasts in the blood or bone marrow (≥20% of nucleated cells). In contrast, MDS patients produce mature blood cells, although maturation and cell morphology are abnormal, and mature cells are reduced in number. Although the block in maturation is a continuum between MDS and secondary AML, higher blast percentage generally confers a worse prognosis in MDS.28,29 A major limitation of using blast count to differentiate MDS from secondary AML is that the blast count is a subjective measurement, relying on a morphological assessment sensitive to interobserver variability.33,34 In addition, MDS blasts can be difficult to distinguish from normal blasts by microscopy, making it difficult to monitor or detect clonal disease in the bone marrow in patients with a normal blast count (eg, <5%). In these cases, the presence of dysplasia (subjective as well) and/or genetic abnormalities is needed to diagnose MDS.
Cell death and proliferation also differ between MDS and secondary AML. Lower-grade MDS is often characterized by increased apoptosis, a noninflammatory cell death that contributes to ineffective myelopoiesis.35-40 Recent research has also linked MDS to increased inflammatory cell death processes, such as pyroptosis and necroptosis.41,42 Unlike apoptosis, these release inflammatory signals and provide a link between cell death and the proinflammatory bone marrow environment observed in MDS patients.41-47 In contrast, secondary AML, and AML in general, is associated with increased cell survival and proliferation. Collectively, the data suggest that the increased cell death phenotype observed in lower-grade MDS gives way to a prosurvival phenotype during progression to higher-grade MDS and secondary AML. Recent work has identified transcriptome, splicing, and DNA methylation alterations in MDS and secondary AML, including in genes associated with hematopoietic cell differentiation, signaling pathways, proliferation, and DNA damage repair. Some of these alterations have been associated with progression to secondary AML and/or clinical outcome.48-51 Future studies using newly developed single-cell techniques (eg, mass cytometer [cytometry by time of flight (CyTOF)], single-cell RNA sequencing) will likely provide additional differences in cell biology between MDS and secondary AML.
The blast count underestimates tumor burden in MDS
MDS was first demonstrated to be a clonal disease by studies of skewed X chromosome inactivation in women,52-54 followed by the identification of recurrent chromosomal abnormalities in up to 50% of MDS patients.55-63 By combining targeted gene panel sequencing with cytogenetics, ≥78% of MDS patients have a clonal genetic abnormality.18,64-67 Using whole-exome sequencing (WES) or whole-genome sequencing (WGS), we now know that nearly all MDS patients have a clonal mutation. Although these genetic studies can identify clonality in MDS, they also provide a measure of tumor burden that often greatly exceeds the bone marrow blast percentage. Indeed, MDS patients with a normal blast count (ie, <5%) can have an abnormal karyotype or mutations in nearly all bone marrow cells using WGS, regardless of the blast count (Figure 1).17,18,68 Although new mutations may not be acquired between high-risk MDS and leukemia, using the blast count to define a boundary between MDS and AML secondary to MDS has limitations, because disease progression is a continuum.69,70 In addition, the presence of specific genetic abnormalities can be diagnostic of leukemia, even with a blast count < 20% (eg, translocations involving RUNX1-RUNX1T1, CBFB-MYH11), providing further support that a blast count cutoff may not always be ideal. Moving forward, incorporating serial sequencing to monitor dynamic changes in MDS tumor burden over time may be useful along with current monitoring criteria, including blast count, to help identify progression earlier, including the emergence and/or expansion of high-risk genetic abnormalities (eg, TP53, RUNX1, and RAS genes).71,72
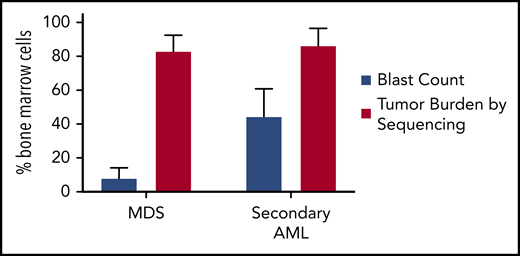
Tumor burden is similar at MDS and secondary AML. Tumor burden at MDS and secondary AML (percentage of bone marrow cells) from 8 patients assessed at both time points.17,18 Tumor burden was measured by morphology using the blast count percentage and sequencing of total bone marrow cells (ie, percentage of clonal cells based on the mutations’ variant allele frequency). Although the blast count increases significantly from MDS to secondary AML, the percentage of clonal cells based on sequencing is similar at both time points. Data are mean ± standard deviation.
Tumor burden is similar at MDS and secondary AML. Tumor burden at MDS and secondary AML (percentage of bone marrow cells) from 8 patients assessed at both time points.17,18 Tumor burden was measured by morphology using the blast count percentage and sequencing of total bone marrow cells (ie, percentage of clonal cells based on the mutations’ variant allele frequency). Although the blast count increases significantly from MDS to secondary AML, the percentage of clonal cells based on sequencing is similar at both time points. Data are mean ± standard deviation.
The genetics of progression from MDS to secondary AML
Genetic abnormalities in MDS and secondary AML
Chromosome abnormalities and structural variants (including copy number alterations [CNAs], inversions, and translocations) are common in MDS and secondary AML. Similar cytogenetic abnormalities are shared between MDS and secondary AML, such as del(5q), del(7q), del(20q), complex karyotype, and others. MDS-associated cytogenetic abnormalities often result in CNAs, as opposed to balanced rearrangements, which are more common in de novo AML [eg, t(15;17), t(8;21), inv(16), and KMT2A rearrangements].58,63,73,74 Additionally, smaller CNAs (<20 kb) and copy number neutral loss of heterozygosity (or uniparental disomy) occur in MDS and secondary AML and can be detected using single-nucleotide polymorphism arrays. CNA and uniparental disomy regions have been reported to harbor driver genes in MDS and secondary AML.75-80 In fact, a set of “MDS-associated” cytogenetic abnormalities is considered diagnostic of the World Health Organization–defined AML-MRC, even in the absence of a prior MDS diagnosis or morphologic dysplasia.3
Large-scale next-generation sequencing studies have consistently identified mutations in genes in ≥6 major cellular pathways that are shared between MDS and secondary AML, including spliceosome genes, epigenetic modifiers, transcription factors, activated signaling genes, cohesin factors, and TP53.17,18,64-67,81-83 In addition to the previously mentioned cytogenetic abnormalities that distinguish secondary AML from de novo AML, the presence of mutations in spliceosome genes (eg, SRSF2, SF3B1, U2AF1), EZH2, BCOR, and STAG2 is highly suggestive that an AML evolved from MDS, even without a known antecedent MDS diagnosis.65,81,84
Although there is a large degree of overlap in the genes mutated in MDS and secondary AML, the frequency of gene mutations is often different between MDS and secondary AML (Figure 2). Mutations in epigenetic modifiers (eg, TET2, DNMT3A) and TP53 are common across a variety of clonal hematopoietic diseases (eg, ARCH/CHIP, MDS, secondary AML, and de novo AML), and splicing factor gene mutations are most common in MDS and secondary AML.66,85,86 This suggests that these mutations occur early in MDS pathogenesis prior to secondary AML progression. In contrast, mutations in transcription factor (eg, RUNX1, GATA2, CEBPA) and activating signaling genes (eg, RAS family genes, FLT3) are more common in secondary AML, suggesting that these mutations are acquired later during disease progression in a subset of cells that expand.49,65,72,81,87-89 Although our understanding of what genes are mutated in MDS and secondary AML has advanced rapidly thanks to next-generation sequencing, a major advantage of this technology is that it can be used to impute tumor clonality and provide insight into clonal evolution.
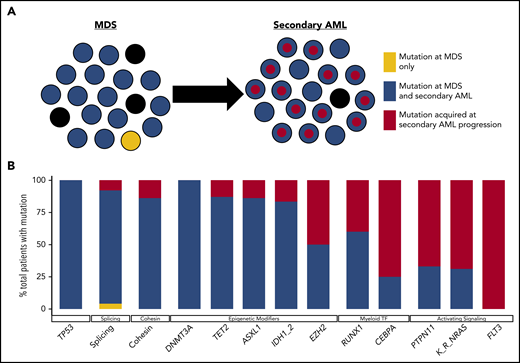
Clonal evolution during progression from MDS to secondary AML. Using previously published paired MDS and secondary AML samples from the same patients (N = 60),17,18,23,65,66,88 the order of mutation acquisition was inferred by assessing the presence or absence of specific mutations at each sampling. (A) A model for sequential accumulation of mutations during progression from MDS to secondary AML. Black cells are normal at MDS and secondary AML. Mutations in blue are acquired early, define the founding clone, and expand to become the most abundant clone in the marrow at MDS diagnosis. These cells then acquire red mutations, form a subclone, and expand at the time of progression to secondary AML. (B) Percentage of patients with a mutation detectable at MDS only (yellow), secondary AML only (red), or at MDS and persisting during disease progression (blue). Mutations in specific functional categories are enriched in 1 of these patterns, with most TP53, epigenetic modifiers, and spliceosome gene mutations present at MDS (eg, 100% of DNMT3A mutations are blue), whereas mutations in transcription factors (eg, RUNX1, CEBPA) and activating signaling genes (RAS family, PTPN11, FLT3) typically expand or are acquired and emerge at progression (eg, 100% of FLT3 mutations are red). This suggests a typical order of mutation acquisition, with blue mutations being acquired early and present in the majority of marrow cells at MDS diagnosis, followed by the red mutations, which expand or are acquired and expand at progression. Only 1 mutation (shown in yellow) that was detected at MDS was not detected at secondary AML (SRSF2 mutation coding for the P95R substitution).88 Adapted from Lindsley et al.65
Clonal evolution during progression from MDS to secondary AML. Using previously published paired MDS and secondary AML samples from the same patients (N = 60),17,18,23,65,66,88 the order of mutation acquisition was inferred by assessing the presence or absence of specific mutations at each sampling. (A) A model for sequential accumulation of mutations during progression from MDS to secondary AML. Black cells are normal at MDS and secondary AML. Mutations in blue are acquired early, define the founding clone, and expand to become the most abundant clone in the marrow at MDS diagnosis. These cells then acquire red mutations, form a subclone, and expand at the time of progression to secondary AML. (B) Percentage of patients with a mutation detectable at MDS only (yellow), secondary AML only (red), or at MDS and persisting during disease progression (blue). Mutations in specific functional categories are enriched in 1 of these patterns, with most TP53, epigenetic modifiers, and spliceosome gene mutations present at MDS (eg, 100% of DNMT3A mutations are blue), whereas mutations in transcription factors (eg, RUNX1, CEBPA) and activating signaling genes (RAS family, PTPN11, FLT3) typically expand or are acquired and emerge at progression (eg, 100% of FLT3 mutations are red). This suggests a typical order of mutation acquisition, with blue mutations being acquired early and present in the majority of marrow cells at MDS diagnosis, followed by the red mutations, which expand or are acquired and expand at progression. Only 1 mutation (shown in yellow) that was detected at MDS was not detected at secondary AML (SRSF2 mutation coding for the P95R substitution).88 Adapted from Lindsley et al.65
Tumor clonality heterogeneity in MDS and secondary AML
WGS of MDS and secondary AML bone marrow samples revealed that these tumors are made up of multiple genetically related, but distinct, clones.17,68 Imputing tumor clonality is possible because there are hundreds of mutations detected using WGS90 and dozens of mutations detected by WES.23,66,88,91 Individual mutations can be clustered based on their variant allele frequency (VAF), with mutations having similar VAFs being present in the same cell or clone.90 The validity of imputing clonality has been confirmed by sequencing single-cell colonies grown from MDS or AML patient bone marrow cells, which showed that sequencing of single-cell–derived colonies recapitulated the imputed clonal architecture in bulk cells.90,92,93 Multiple studies have shown that a patient’s bone marrow sample can be clonally complex and contain multiple genetically related, but unique, clones, including a founding clone and subclone(s) derived from the founding clone.17,18,26,66,68,88,93,94 Imputing accurate clonal architecture is limited when mutations are present at low VAFs, making it challenging to determine whether mutations co-occur in the same cell or exist in parallel subclones. In these cases, single-cell sequencing technologies may be needed; however, they can be limited by allele drop-out.
The combination of mutations that occurs within a patient or clone is not necessarily random, with some mutations co-occurring at frequencies greater (or less) than expected by chance. For example, the combination of ASXL1 and U2AF1 has been reported at a greater-than-expected rate, as has STAG2 with several mutations (including RUNX1, SRSF2, and EZH2).64,95,96 Combinations go beyond mutations alone (eg, SF3B1 mutations are associated with overexpression of EV11, including some with EVI1 rearrangements).49 In contrast, mutations in splicing factor genes are rarely comutated, and the same is seen with cohesin complex genes.19,67,83,97,98 The lack of co-occurrence of these mutations may be driven by functional complementarity/redundancy or synthetic lethality.99 Together, these studies demonstrate that MDS and secondary AML tumors are made up of multiple clones, each containing multiple mutations that likely cooperate to cause the disease phenotype.
The frequency of mutations in epigenetic modifier genes in MDS and secondary AML suggests that epigenetic alterations and heterogeneity may play a significant role in disease progression. Although outside the scope of this review, this topic has been covered in recent reviews.86,100,101 Further work in this area may provide new information regarding how genetic and nongenetic abnormalities affect cell phenotypes and could provide insight into effective treatments.
Order of mutation acquisition
Although the co-occurrence of gene mutations is important, the order of mutation acquisition is likely critical for disease pathogenesis and may also have an important impact on the design of clinical trials using targeted therapies. Deciphering the exact order of mutation acquisition in a patient can be difficult. We can gain insight into this question based on several observations. Recent studies have shown that up to 10% of people older than 70 years of age have clonal hematopoiesis, defined by somatic mutations in their blood cells, and are classified as having ARCH/CHIP.13,14,20,21,25,102,103 Many of these mutations (eg, DNMT3A, TET2, ASXL1, and spliceosome genes) are common, with high VAFs in MDS and secondary AML, suggesting that mutations in these genes occur early in the disease pathogenesis and are present in the majority of cells.17,18,64-67,81,82 Consistent with this, mutations in these genes occur in the founding clone of MDS and secondary AML patients, whereas transcription factor and signaling gene mutations typically occur in subclones derived from the founding clone or other subclones (Figure 2).17,23,24,65,66,68 By sequencing a large number of low- and high-risk MDS samples, as well as secondary AML patients, Makishima et al identified 2 classes of genes that were mutated: type 1 (enriched in secondary AML compared with high-risk MDS) and type 2 (enriched in high-risk compared with low-risk MDS).66 Mutations in these genes suggest a typical order of mutation acquisition during progression. However, our understanding of the order of mutation acquisition remains incomplete, because most data are based on the frequency of gene mutations in cohorts of unpaired MDS and secondary AML patients rather than serial samples from the same patient.
To address this limitation, we identified 60 patients in the literature with paired serial MDS/secondary AML samples (Figure 2).17,18,23,65,66,88 Mutations in common ARCH/CHIP-associated genes (eg, TP53, spliceosome, and epigenetic modifiers) detected at secondary AML were already present in the MDS sample (100%, 92%, and 84% of mutations were present at MDS, respectively) with high average VAFs (33%, 43%, and 31%, respectively), indicating that they are acquired early in the disease course. In contrast, mutations in signaling genes were less commonly detected at MDS (34% of mutations present at MDS), indicating that they are gained later in disease progression (Figure 2).17,18,23,65,66,88 The wide range of sequencing approaches used across these studies limited the ability to consistently detect low-level mutations and define clonality. However, these studies do provide important evidence that a typical order of mutation acquisition may exist during progression from MDS to secondary AML. Given that the order of mutation acquisition has been shown to be important for the hematopoietic phenotype in myeloproliferative neoplasms,104,105 the specific order of mutation acquisition may be important for progression from MDS to secondary AML. Future studies using WGS coupled with ultradeep error-corrected sequencing of somatic mutations from a larger number of paired MDS and secondary AML serial samples from the same patient could help to address these questions.
Subclone expansion defines disease progression
In addition to elucidating the clonality of MDS and secondary AML, WGS and other sequencing platforms have revealed that the expansion of a subclone is a common feature of progression from MDS to secondary AML (Figure 3). The expansion of subclones during progression can occur over a relatively short period of time (eg, weeks to months).17,18 However, some subclones that become the most abundant clone at secondary AML can be detected at low levels in MDS months to years prior to progression, suggesting that their presence may be prognostic for future progression.17,23,66,82,88,106 Indeed, subclonal driver mutations are an independent risk factor for disease progression in chronic lymphocytic leukemia.107 Which gene mutations preexist at MDS will also likely matter. Initial studies by Bejar et al identified mutations (eg, RUNX1, TP53, EZH2) that were independent predictors of poor outcome for MDS, although not necessarily disease progression.71 The presence of rare RAS family member gene mutations at MDS was shown to have the same poor outcome regardless of VAF, suggesting that the presence of these mutations is important.72 Consistent with this, several studies with paired MDS and secondary AML samples have shown that signaling gene mutations, along with new cytogenetic abnormalities, expand at the time of disease progression.17,23,88
![Patterns of clonal evolution during the progression of MDS to secondary AML. Multiple patterns of subclone expansion are associated with progression from MDS to secondary AML. Subclones (red, green) can be acquired from the founding clone (blue) in a sequential (ie, linear) order (A) or in parallel (ie, branching) (B). Clonal evolution can also be influenced by treatment, including transplant and chemotherapy. (C) Although chemotherapy can suppress the founding clone [eg, lenalidomide in del(5q)-associated MDS], the acquisition of additional mutations (eg, TP53) or cytogenetic abnormalities can occur during disease progression and contribute to subclone expansion. Similar patterns of progression can occur following progression after a transplant. (D) Treatment may also cause subclone clearance while sparing the founding clone (eg, MEK inhibitor repressing a RAS-mutated subclone). Progression can occur when a new subclone emerges carrying additional mutations (green) that drive progression to secondary AML. allo-HSCT, allogeneic hematopoietic stem call transplant; CR, complete remission. Adapted from Nangalia et al.108](https://ash.silverchair-cdn.com/ash/content_public/journal/blood/136/1/10.1182_blood.2019000942/4/m_bloodbld2019000942cf3.png?Expires=1771329136&Signature=V7Vhi-3WxkztSJzBfkAc0-GW-GahNzdegBqh~oCjStyFH7ONhAqiG4PE3V1hd8D8v59KEemG7jigPya0cuUI~xULrqi3qzJry3bN9iyrdGleATfRgObFLy~Yy7qkaXG8FEiTbtDEQmlY8NSZyO4A~9yggfWNCINQqEfuRryoeRWUjxcWwp-6WRHSpEYQLrlR754xzJaTtAjS72An48YlDvbxtHkN1dq6wDfAf10McepQvTwG4ON03sxPk9ppRf~YJ~ydgzaolECQZz-2ttbq4fCIcFgBdtwXTFbfY9TrCiVynRBMJFffoZ155ZWQO6BCzPEWc44xYQs41Iv3RjGCyQ__&Key-Pair-Id=APKAIE5G5CRDK6RD3PGA)
Patterns of clonal evolution during the progression of MDS to secondary AML. Multiple patterns of subclone expansion are associated with progression from MDS to secondary AML. Subclones (red, green) can be acquired from the founding clone (blue) in a sequential (ie, linear) order (A) or in parallel (ie, branching) (B). Clonal evolution can also be influenced by treatment, including transplant and chemotherapy. (C) Although chemotherapy can suppress the founding clone [eg, lenalidomide in del(5q)-associated MDS], the acquisition of additional mutations (eg, TP53) or cytogenetic abnormalities can occur during disease progression and contribute to subclone expansion. Similar patterns of progression can occur following progression after a transplant. (D) Treatment may also cause subclone clearance while sparing the founding clone (eg, MEK inhibitor repressing a RAS-mutated subclone). Progression can occur when a new subclone emerges carrying additional mutations (green) that drive progression to secondary AML. allo-HSCT, allogeneic hematopoietic stem call transplant; CR, complete remission. Adapted from Nangalia et al.108
Patterns of clonal evolution during the progression of MDS to secondary AML. Multiple patterns of subclone expansion are associated with progression from MDS to secondary AML. Subclones (red, green) can be acquired from the founding clone (blue) in a sequential (ie, linear) order (A) or in parallel (ie, branching) (B). Clonal evolution can also be influenced by treatment, including transplant and chemotherapy. (C) Although chemotherapy can suppress the founding clone [eg, lenalidomide in del(5q)-associated MDS], the acquisition of additional mutations (eg, TP53) or cytogenetic abnormalities can occur during disease progression and contribute to subclone expansion. Similar patterns of progression can occur following progression after a transplant. (D) Treatment may also cause subclone clearance while sparing the founding clone (eg, MEK inhibitor repressing a RAS-mutated subclone). Progression can occur when a new subclone emerges carrying additional mutations (green) that drive progression to secondary AML. allo-HSCT, allogeneic hematopoietic stem call transplant; CR, complete remission. Adapted from Nangalia et al.108
Sequencing serial samples is critical to define expanding subclones. Analysis of clonal evolution has shown that evolution can occur in 1 of 2 patterns: linear, in which each successive subclone occurs within the previous parental clone, or branched, in which a founding clone can generate several parallel subclones (Figure 3A-B).17,23,66,68,88,108 In other cases, it has been shown that a subclone can expand to occupy the entire tumor, forcing the collapse of other subclones, a process termed “clone sweeping.”66 WGS of additional paired MDS and secondary AML samples, capable of identifying hundreds of mutations in each sample, will help to define the patterns of clonal evolution (clone collapse and expansion) associated with disease progression. Identifying the genetic patterns of clonal expansion during disease progression may provide important information regarding genetic drivers of subclone expansion during progression to secondary AML.
Clonal diversity of hematopoietic stem cells in MDS and secondary AML
MDS and secondary AML are diseases of hematopoietic stem cells,109-114 with somatic mutations being present in myeloid and lymphoid cells from MDS patients.88,115,116 This suggests that the hematopoietic stem cell, myeloid biased or otherwise, is the cell of origin of disease. Recently, Chen et al sequenced a limited number of phenotypically defined stem cell populations in MDS/secondary AML patients using cell sorting, followed by whole-genome amplification and targeted sequencing.26 The results indicate that subclonal diversity may be higher in stem cells than in blasts at MDS and secondary AML. Furthermore, they show that the dominant clone in the stem cells at MDS (ie, the clone of stem cells giving rise to blasts) is not always the same as the dominant clone at AML, indicating a potential nonlinear path of clonal evolution at the stem cell level. These results indicate a possible greater level of clonal diversity than appreciated using bulk bone marrow samples. These data raise interesting questions regarding the clonal diversity of stem cells in MDS and secondary AML and warrant further study.
Effects of MDS treatment on the clonal evolution of secondary AML
As outlined above, clonal evolution during progression to secondary AML is characterized by the persistence of founding clone mutations and, typically, the expansion of a subclone with unique mutations. Although this pattern is similar for patients receiving treatment or supportive care (transfusions, erythropoietin, thrombopoietin receptor agonists, or granulocyte colony-stimulating factor),17,23,68 several studies have addressed whether specific therapies influence the pattern of clonal evolution.
Clonal evolution following lenalidomide treatment in patients with del(5q) MDS
One of the most common chromosomal abnormalities in MDS/secondary AML is del(5q).74 These patients can achieve a complete remission following lenalidomide treatment.117,118 Although lenalidomide is associated with clinical responses and a decrease in clonal del(5q) cells in the marrow, del(5q) was shown to persist in the quiescent stem cell population (CD34+/CD38−/CD90+).112 These patients eventually relapse, with an expansion of the del(5q) cells containing additional abnormalities (eg, TP53 mutations) (Figure 3C).23,112 Consistent with these results, TP53 mutations were present at low levels months to years prior to disease progression in del(5q)-associated MDS.119 These studies demonstrate that selective pressure from chemotherapy can impact clonal evolution during progression to secondary AML.
Effect of hypomethylating agents on disease progression
Using DNA-hypomethylating agents (HMAs) to treat higher-risk MDS or older AML patients is common.120,121 Although this represents a noncurative therapy, HMAs are associated with improved overall survival.121-124 Predicting response to HMAs based on a patient’s gene mutations remains challenging, and results are variable across studies. Various studies have identified that MDS patients with mutations in epigenetic modifiers (eg, TET2, DNMT3A) and MDS and AML patients with complex cytogenetics and/or TP53 mutations respond better to decitabine.82,125-129 Of note, HMA therapy has been shown to induce C>G transversions in vitro, and is associated with C>G transversions in MDS and AML cells from patients, indicating that HMAs may influence clonal evolution.82,106,130 These C>G transversion mutations can occur in genes implicated in disease pathogenesis, as well as in subclones that emerge at relapse, raising questions of whether HMAs may induce pathogenic mutations.106 Previous studies of decitabine-treated patients utilizing fluorescent in situ hybridization and flow-sorted hematopoietic stem and progenitor cells showed that genetic relapse can predate morphological relapse,113,131 providing the rationale to incorporate sequencing to monitor tumor burden in patients. Tracking all clones will be important, because some patients have a reduction in a subclone with no change in the size of the founding clone (Figure 3D).82 Future studies are needed to define how to incorporate serial sequencing results into traditional response criteria to monitor tumor burden and detect expanding clones.
Disease progression following bone marrow transplantation for MDS and secondary AML
The only curative therapy for MDS and secondary AML is allogeneic hematopoietic stem cell transplant (HSCT). For patients who qualify, most often those with intermediate-2 or higher risk by the International Prognostic Scoring System who are younger than 75 years and in good health with minimal comorbidities,132 HSCT can improve outcomes, although relapse is still common. The use of reduced-intensity conditioning regimens has increased the number of older MDS patients receiving an HSCT.133,134 The pattern of clonal evolution and disease progression following transplant shares some similarities with postchemotherapy progression (Figure 3C). A study of 9 MDS patients who progressed following transplant showed that subclones emerged in 8 of 9 patients at progression, often harboring a new structural variant.106 Furthermore, founding clone mutations were detectable at relapse in all cases, with many mutations being detectable as early as 30 and/or 100 days posttransplant.106 These results suggest that monitoring patients for molecular residual disease may identify a high-risk group of individuals who could be considered for an early intervention in a clinical trial.
Monitoring disease progression
What is the optimal approach to monitor MDS patients for disease progression? Flow cytometry is able to identify dyspoiesis in MDS patients that correlates with clinical outcomes and genetic abnormalities.135-139 The utility of flow cytometry in the clinical work-up of MDS patients has been outlined previously by the European LeukemiaNet Working Group.140,141 Thus, along with morphology, flow cytometry provides a tool for assessment and monitoring of MDS patients. Given the prevalence of genetic abnormalities in these patients, incorporating sequencing results into traditional response criteria may allow for better tumor burden monitoring, particularly in patients with a normal karyotype (Figure 1).17,18,82,91,106,142,143 However, this will have to be tested in prospective clinical trials that use International Working Group response criteria, serial sequencing (for mutation VAFs and clones), and outcomes including survival.144 The optimal sequencing approach to monitor tumor burden and disease MDS progression remains an open question. There are advantages and limitations for various sequencing platforms that could influence how they get incorporated in the clinic.
Whole-genome sequencing
WGS can detect hundreds of mutations per sample, making it ideal for imputing tumor clonality17,18,68,92,93 ; however, sequencing depth is often limited by costs, making it difficult to detect mutations with VAFs < 10%. Therefore, low-level subclones can be missed. Additionally, sequencing of paired normal DNA is necessary to confidently identify somatic mutations (Figure 4). Despite these limitations, in situations in which the goal is to define the clonal architecture of a sample or discover structural variants and mutations in noncoding regions of the genome, WGS remains an ideal approach.
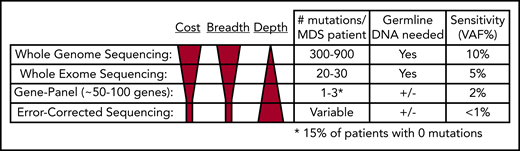
Comparison of next-generation sequencing platforms. The optimal sequencing platform to use for clinical testing is dependent on several variables, include the cost, breadth of sequencing coverage (ie, the number of mutations that can be detected), and the typical sequencing depth obtained (ie, the sensitivity of variant detection). Although WGS provides the greatest breadth, the standard depth of coverage (30-45×) limits detection of variants to those with a VAF typically >10%. WES only provides coverage of coding bases in the genome, but the greater sequencing depth (typically 75-150×) allows for detection of mutations with VAFs as low as 5%. Gene panel sequencing is limited to the most commonly mutated genes but can detect mutations at lower VAFs. A typical 100-gene panel with 1000× coverage depth can detect mutations with VAFs as low as 2%, at a reasonable cost. Finally, error-corrected sequencing provides extremely high-depth coverage (10 000×) along with error correction via unique molecular indexes, together allowing for detection of VAFs < 1%. Error-corrected sequencing and gene panel sequencing approaches offer a large degree of flexibility, because they can be used to validate mutations (ie, those detected by WGS), track mutations, or discover mutations. In addition, these methods are often used without paired normal DNA to detect variants. However, germline DNA is necessary to definitively identify somatic mutations (+/−). Adapted from Jacoby et al.90
Comparison of next-generation sequencing platforms. The optimal sequencing platform to use for clinical testing is dependent on several variables, include the cost, breadth of sequencing coverage (ie, the number of mutations that can be detected), and the typical sequencing depth obtained (ie, the sensitivity of variant detection). Although WGS provides the greatest breadth, the standard depth of coverage (30-45×) limits detection of variants to those with a VAF typically >10%. WES only provides coverage of coding bases in the genome, but the greater sequencing depth (typically 75-150×) allows for detection of mutations with VAFs as low as 5%. Gene panel sequencing is limited to the most commonly mutated genes but can detect mutations at lower VAFs. A typical 100-gene panel with 1000× coverage depth can detect mutations with VAFs as low as 2%, at a reasonable cost. Finally, error-corrected sequencing provides extremely high-depth coverage (10 000×) along with error correction via unique molecular indexes, together allowing for detection of VAFs < 1%. Error-corrected sequencing and gene panel sequencing approaches offer a large degree of flexibility, because they can be used to validate mutations (ie, those detected by WGS), track mutations, or discover mutations. In addition, these methods are often used without paired normal DNA to detect variants. However, germline DNA is necessary to definitively identify somatic mutations (+/−). Adapted from Jacoby et al.90
Whole-exome sequencing
In contrast to WGS, WES typically only detects dozens of mutations in MDS and secondary AML samples; however, greater sequencing depths can be achieved at a reasonable cost using WES, allowing detection of mutation VAFs as low as 5%.23,66,88,91 Germline DNA is still required to confidently identify somatic mutations (Figure 4). Although most patients have a detectable coding mutation, it remains challenging to accurately define clones; however, retrospective WES studies have shown that subclones that expand at progression can be detected prior to progression.66,82,105 If clinically relevant subclonal mutations could be identified in real time, it may be possible to implement a clinical intervention to prevent progression.
Gene-panel sequencing
Targeted gene-panel sequencing is widely available in the clinic.18,64,66,67,97 Gene panels of ∼100 genes or mutation hotspots are able to detect a mutation in most patients (Figure 4); however, gene panel sequencing is limited by the fact that a subset of patients will not have a detectable mutation. Two large sequencing studies interrogated 944 and 738 MDS patients for mutations in 104 and 111 genes, respectively.64 Mutations were identified in 845 (89.5%) and 549 (74%) patients, respectively.64,67 Similar findings were reported for secondary AML; a panel covering 40 genes identified a mutation in 90 of 93 (97%) patients.65 A major limitation of gene panel sequencing is that too few mutations are identified to confidently identify clones, with many clones being completely missed if they do not harbor a mutation in one of the sequenced genes.
Error-corrected sequencing
An error-corrected sequencing (ECS) approach is based on using unique molecular identifiers coupled with high-sequencing coverage depths.82,91,106,145,146 ECS allows for very low detection of a designated set of mutations (<1% VAF). It can be used in 2 ways: as a gene panel to discover mutations or to validate and track previously identified mutations (ie, by WGS) (Figure 4). ECS may provide significant utility in the setting of residual disease detection. In the transplant setting, residual disease detection has clinical relevance when detected by morphology, quantitative polymerase chain reaction, chimerism analysis, and flow cytometry147-151 ; however, these technologies have limitations. In an exploratory study of MDS patients who received allogeneic HSCT, Duncavage et al found that detection of persistent mutations at VAF ≥ 0.5% at day 30 posttransplant was associated with increased disease progression and lower progression-free survival.91 Patients had a median of 67 days between day-30 mutation detection and disease progression,91 a period that may allow for salvage therapy in some patients.152-156 Kim et al also observed that detecting mutations posttransplant for de novo AML impacts outcomes.157
Together, the benefits and limitations of these sequencing technologies indicate that the best approach is dependent on the specific clinical question being asked. The optimal sequencing technique will depend on whether the goal is diagnostic, the identification of clones, or the monitoring of residual disease. It will be critical to test the feasibility and utility of each application in well-controlled prospective clinical trials before implementation in the clinic becomes standard.
Concluding thoughts
The introduction of next-generation sequencing for MDS and secondary AML confirms that clonal evolution accompanies disease progression, as previously observed using cytogenetics. These studies show that MDS and secondary AML represent a disease continuum that genetically evolves, rather than a new disease. Sequencing results have consistently shown that the blast count assessment, although an important risk factor, often underestimates the tumor burden in MDS. Based on our current knowledge, we suggest that it is time to augment our clinical assessment of tumor burden based on morphology with next-generation sequencing data and develop an approach to incorporate molecular-based disease classification and tumor burden to better diagnose and monitor MDS. Future studies, especially ones involving paired MDS and secondary AML samples from the same patient, could provide insight into a critical conserved order of mutation acquisition that drives disease pathogenesis and is predictive of MDS progression, potentially allowing for early intervention.
Acknowledgments
The authors thank Meagan A. Jacoby and Eric J. Duncavage for helpful scientific discussions and Sridhar N. Srivatsan for assistance with figures.
This work was supported by National Institutes of Health, National Cancer Institute (grants P01 CA101937 and R33CA217700), the Edward P. Evans Foundation, and the Lottie Caroline Hardy Trust (all M.J.W.).
The authors apologize to investigators whose work could not be included because of space constraints.
Authorship
Contribution: A.J.M. and M.J.W. jointly wrote the manuscript.
Conflict-of-interest disclosure: The authors declare no competing financial interests.
Correspondence: Matthew J. Walter, Washington University School of Medicine, 660 S. Euclid Ave, Campus Box 8007, St. Louis, MO 63110; e-mail: mjwalter@wustl.edu.

This feature is available to Subscribers Only
Sign In or Create an Account Close Modal