

Key Points
NETs contribute to microthrombi through platelet-neutrophil interactions in COVID-19 ARDS.
nNIF blocks NETs induced by COVID-19 plasma and represents a potential therapeutic intervention in COVID-19.

Abstract
COVID-19 affects millions of patients worldwide, with clinical presentation ranging from isolated thrombosis to acute respiratory distress syndrome (ARDS) requiring ventilator support. Neutrophil extracellular traps (NETs) originate from decondensed chromatin released to immobilize pathogens, and they can trigger immunothrombosis. We studied the connection between NETs and COVID-19 severity and progression. We conducted a prospective cohort study of COVID-19 patients (n = 33) and age- and sex-matched controls (n = 17). We measured plasma myeloperoxidase (MPO)-DNA complexes (NETs), platelet factor 4, RANTES, and selected cytokines. Three COVID-19 lung autopsies were examined for NETs and platelet involvement. We assessed NET formation ex vivo in COVID-19 neutrophils and in healthy neutrophils incubated with COVID-19 plasma. We also tested the ability of neonatal NET-inhibitory factor (nNIF) to block NET formation induced by COVID-19 plasma. Plasma MPO-DNA complexes increased in COVID-19, with intubation (P < .0001) and death (P < .0005) as outcome. Illness severity correlated directly with plasma MPO-DNA complexes (P = .0360), whereas Pao2/fraction of inspired oxygen correlated inversely (P = .0340). Soluble and cellular factors triggering NETs were significantly increased in COVID-19, and pulmonary autopsies confirmed NET-containing microthrombi with neutrophil-platelet infiltration. Finally, COVID-19 neutrophils ex vivo displayed excessive NETs at baseline, and COVID-19 plasma triggered NET formation, which was blocked by nNIF. Thus, NETs triggering immunothrombosis may, in part, explain the prothrombotic clinical presentations in COVID-19, and NETs may represent targets for therapeutic intervention.
Introduction
SARS-CoV-2 infection is now confirmed in nearly 12 million individuals worldwide, and COVID-19 has caused >540 000 fatalities globally.1 As the pandemic continues, evidence suggests that microthrombi and coagulopathy contribute to COVID-19 pathogenesis and varied disease presentations. Up to 10% of all COVID-19 patients will become critically ill with multiorgan failure and require admission to the intensive care unit (ICU) for acute respiratory distress syndrome (ARDS), putting an unprecedented strain on health care systems.2 Successfully meeting the clinical challenge of COVID-19 will require new insights into disease pathogenesis and novel treatments for COVID-19 patients. Immunothrombosis, the direct interaction of activated leukocytes with platelets and plasma coagulation factors in the innate immune response, may contribute to the thrombotic events described in COVID-19 patients with coagulopathy.3 Furthermore, such pathogenic immunothrombosis may result from dysregulated neutrophil extracellular trap (NET) formation in COVID-19.4 Thus, dismantling or inhibiting NETs in COVID-19 could ameliorate NET-mediated inflammatory and thrombotic tissue damage associated with COVID-19 ARDS and death.4
Polymorphonuclear leukocytes (PMNs; neutrophils) produce NETs through a regulated cell death process termed NETosis.5 NETs are extracellular 3-dimensional lattices of decondensed chromatin decorated with histones and antimicrobial proteins that are released upon stimulation. Pathogens, including respiratory viruses, induce NETosis, leading to NETs that physically trap and kill microbes as part of the innate immune system.6 When triggered by platelets, NETosis can become dysregulated leading to NET-mediated tissue damage, hypercoagulability, and thrombosis.7 Dysregulated NETosis is associated with acute and chronic inflammatory disease.8 The contribution of NETosis to sepsis and ARDS pathogenesis is well documented, with NETs causing vascular tissue damage and scattered microthrombi leading to multiorgan failure and death.9-13
The most common complications of COVID-19 include ARDS, sepsis, and multiorgan dysfunction syndrome associated with high morbidity and mortality.14,15 Similar to other infectious syndromes, the tissue injury seen in SARS-CoV-2 infection is driven by a dysregulated immune response, release of inflammatory cytokines, and development of pathogenic microvascular thrombi.16 In this study, we provide clinical evidence that increased NET formation correlates with COVID-19–related ARDS and is a potential biomarker for disease severity. We also demonstrate that COVID-19 patients have increased plasma platelet factor 4 (PF4) and RANTES, which are known to trigger NETosis.17 We also provide evidence that neonatal NET-inhibitory factor (nNIF), a NET-inhibitory peptide discovered in human umbilical cord blood,18,19 inhibits NET formation induced by plasma from COVID-19 patients. We believe that these findings have broad relevance across the acute inflammatory lung disease spectrum and are not necessarily specific to COVID-19.
Patients and methods
Study design
In this investigator-initiated, prospective cohort study, we enrolled 33 COVID-19 patients and 17 age- and sex-matched healthy adults (Table 1) admitted to the University of Utah Health Sciences Center with respiratory symptoms and positive SARS-CoV-2 polymerase chain reaction (PCR) testing. We investigated elements implicated in the pathogenesis of COVID-19–related sepsis, ARDS, and thrombosis, including NET formation and platelet-neutrophil interactions. We conducted all in vitro experiments within our laboratory’s BioSafety Level 2 Plus facility with appropriate personal protective equipment, N-95 respirators for work with COVID-19 tracheal aspirate samples, and confocal microscopy only after cell and supernatant fixation.
Clinical characteristics of healthy donors and hospitalized patients with COVID-19
| Characteristics | Healthy donors (n = 17) | Hospitalized | P | |
|---|---|---|---|---|
| Non-ICU COVID-19 infection (n = 19) | ICU COVID-19 infection (n = 14) | |||
| Age, mean ± SD, y | 50.6 ± 17.6 | 48.2 ± 13.6 | 64.5 ± 13.7 | .008 |
| Male | 50 | 52.6 | 57.1 | .94 |
| Hispanic/Latino/African American | 11.8 | 31.6 | 42.9 | .15 |
| BMI (kg/m2), mean ± SD | 33.9 ± 9.6 | 30.5 ± 9.4 | .43 | |
| Diabetes | 31.6 | 57.1 | .15 | |
| Hypertension | 36.8 | 42.9 | .74 | |
| Chronic lung disease | 26.3 | 42.9 | .33 | |
| SOFA score, mean ± SD | 1.6 ± 1.3 | 4.6 ± 1.2 | <.0001 | |
| ARDS | 10.5 | 92.9 | <.0001 | |
| Mechanical ventilation | 0.0 | 50.0 | <.0001 | |
| 28-d survival | 100 | 71.4 | .011 | |
| WBC (k/uL), mean ± SD | 6.1 ± 2.4 | 8.3 ± 2.3 | .02 | |
| Platelet count (k/uL), mean ± SD | 245 ± 107 | 244 ± 56 | .97 | |
| Characteristics | Healthy donors (n = 17) | Hospitalized | P | |
|---|---|---|---|---|
| Non-ICU COVID-19 infection (n = 19) | ICU COVID-19 infection (n = 14) | |||
| Age, mean ± SD, y | 50.6 ± 17.6 | 48.2 ± 13.6 | 64.5 ± 13.7 | .008 |
| Male | 50 | 52.6 | 57.1 | .94 |
| Hispanic/Latino/African American | 11.8 | 31.6 | 42.9 | .15 |
| BMI (kg/m2), mean ± SD | 33.9 ± 9.6 | 30.5 ± 9.4 | .43 | |
| Diabetes | 31.6 | 57.1 | .15 | |
| Hypertension | 36.8 | 42.9 | .74 | |
| Chronic lung disease | 26.3 | 42.9 | .33 | |
| SOFA score, mean ± SD | 1.6 ± 1.3 | 4.6 ± 1.2 | <.0001 | |
| ARDS | 10.5 | 92.9 | <.0001 | |
| Mechanical ventilation | 0.0 | 50.0 | <.0001 | |
| 28-d survival | 100 | 71.4 | .011 | |
| WBC (k/uL), mean ± SD | 6.1 ± 2.4 | 8.3 ± 2.3 | .02 | |
| Platelet count (k/uL), mean ± SD | 245 ± 107 | 244 ± 56 | .97 | |
Unless otherwise noted, data are percentages (%).
BMI, body mass index; SOFA, Sequential Organ Failure Assessment; WBC, white blood cell count.
Study patients
We collected 60 mL of anticoagulant citrate dextrose-anticoagulated whole blood from patients hospitalized with COVID-19 at the University of Utah Health Science Center from 17 March 2020 to 30 May 2020. All COVID-19 patients were identified under study protocols approved by the Institutional Review Board (IRB) of the University of Utah (IRB 00102638 and 00093575). Healthy blood donors enrolled under a separate IRB protocol (IRB 0051506). Each study participant or their legal authorized representative gave written informed consent for study enrollment in accordance with the Declaration of Helsinki. Enrollment criteria included age >18 years, respiratory symptoms (cough, shortness of breath) or fever, hospital admission, positive SARS-CoV-2 PCR testing, and informed consent. Demographic and illness severity data are summarized in Table 1. We also enrolled 3 convalescing COVID-19 patients outside of our cohort who were not hospitalized and 2 convalescing patients from our cohort to collect blood samples from 4 to 6 weeks after they tested positive for SARS-CoV-2.
MPO-DNA ELISA
We detected myeloperoxidase (MPO)-DNA complexes as a measure of NETs in plasma and available tracheal aspirate samples using the MPO-DNA enzyme-linked immunosorbent assay (ELISA).20 We used an anti-human MPO primary antibody (Upstate Biotechnology) as the capture antibody and a peroxidase-labeled anti-DNA primary antibody (Cell Death Detection ELISA Kit; Roche) as the detection antibody. Plasma and tracheal aspirate samples were diluted 1:3 and 1:15 with phosphate-buffered saline, respectively. Results are reported as the percentage of healthy adult plasma ± SD, arbitrarily set at 100%.
PMN isolation
We isolated PMNs from freshly collected whole blood of healthy adults and COVID-19 patients using an EasySep Direct Human Neutrophil Isolation Kit (STEMCELL Technologies) with >95% purity. We resuspended the PMNs to a concentration of 2 × 106 cells per milliliter in serum-free M-199 and performed all of our experiments at 37°C in 5% CO2/95% air.
Neutrophil granularity quantification
Neutrophils from COVID-19 patients (n = 4) and healthy donors (n = 6) were stained with anti-human CD66b-V450 antibodies (1:20, 560861; BD Biosciences) for 15 minutes at 37°C in the dark and fixed with FACS lysis buffer. Granularity was assessed by side scatter area using a CytoFLEX Flow Cytometer (Becton Dickinson).
NET imaging and quantification
We assessed human NET formation as previously described.18 We treated PMNs isolated from healthy adults and COVID-19 patients with phorbol 12-myristate 13-acetate (PMA; 20 nM; 2 hours), polyinosinic-polycytidylic acid [poly(I:C); 1 μM; 2 hours], or COVID-19 patient plasma (undiluted; 2 hours), with and without a 1-hour pretreatment with nNIF or its inactive scrambled peptide control (1 nM). For all experiments, we visualized NET formation using confocal microscopy (Olympus) with cell-permeable (SYTO Green) or cell-impermeable (SYTOX Orange; both from Molecular Probes) DNA fluorescent dyes. We quantitatively assessed NET formation using a cell-free DNA fluorescence assay with SYTOX Green DNA dye. Relative fluorescence was quantified using a fluorometric plate reader with SoftMax software (Molecular Devices).21 Given the challenges of working in a Biosafety Level 2 Plus facility, our in vitro studies of NET formation relied on the combination of confocal imaging with representative images and a high-throughput cell-free DNA fluorescence assay quantitation.
Immunofluorescence staining for NETs and platelets
Paraffin-embedded autopsy lung specimens from 3 cases of COVID-19 (Weill Cornell Medical Center) were stained largely as previously described.22 Commercially available healthy lung tissue (US Biomax; NCT086) served as a control. The tissue sections available were only 4 to 5 μm thick, making detection of extensive lattices of extracellular NETs difficult. Briefly, to stain for neutrophils and NETs, we used mouse anti-human MPO (1:400, MAB3174; R&D Systems) and rabbit anti-human citrullinated histone H3 (1:250, ab5103; Abcam) antibodies. To stain for NETs and PF4, we used anti-citrullinated histone H3 and mouse anti-human PF4 (1:200sc-398979; Santa Cruz Biotechnology) antibodies with Alexa Fluor 488–conjugated anti-mouse and Alexa Fluor 568–conjugated anti-rabbit secondary antibodies, blocked using a M.O.M. (Mouse on Mouse) Immunodetection Kit, Basic (BMK-2202; Vector Laboratories), followed by incubation with anti-MPO antibodies and Alexa Fluor 647–conjugated anti-mouse antibody. We used 4′,6-diamidino-2-phenylindole (DAPI) as a nuclear counterstain (Thermo Fisher Scientific). Images were captured using confocal microscopy (Leica SP8) and analyzed using Volocity software (version 6.3.0; PerkinElmer).
Plasma coagulation factor assays
We used ELISA kits to quantify soluble PF4 and RANTES (R&D Systems) in plasma from COVID-19 patients and healthy donors. We used additional ELISA kits (Abcam) to quantify von Willebrand factor (VWF) antigen and D-dimers in the same samples.
Platelet-neutrophil aggregates
We diluted whole blood 1:10 with Tyrode’s/HEPES buffer and labeled platelets with anti-human CD41-APC antibodies (1:50; 559777) and PMNs with anti-human CD66b-V450 antibodies (1:20; 560861; both from BD Biosciences) for 15 minutes at 37°C in the dark. We fixed them with FACS lysis buffer and determined fluorescence using a CytoFLEX Flow Cytometer (Becton Dickinson).
Cytokine array
We measured cytokine levels in equal volumes of healthy adult and COVID-19 patient plasma using a multiplex bead array for interleukin-8 (IL-8) and IL-6 (MilliporeSigma), according to the manufacturer’s instructions, and analyzed them on a Luminex 200 machine.
nNIF peptide synthesis
nNIF and its inactive scrambled peptide control were synthesized, as previously described, by the University of Utah DNA/Peptide Synthesis Core Facility.18
Statistical analysis
For comparisons of the hospitalized patient groups, we determined the mean ± standard deviation (SD). For each in vitro experimental variable, we determined the mean ± standard error of the mean (SEM). For group comparisons, we used an unpaired Student t test or a 1-way analysis of variance (ANOVA) with Tukey’s multiple-comparisons post hoc testing, as appropriate (GraphPad Prism v8.4). Data that were not distributed normally were analyzed with a Mann-Whitney statistical test for pairwise comparisons. Correlations were calculated using Spearman’s rank correlation. All data used in each statistical test met the assumptions of the specific test. All tests were 2 sided, and P < .05 was considered statistically significant.
Results
Increased plasma NETs correlate with increased COVID-19 severity
We assessed NETs in autopsy lung specimens from 3 COVID-19 patients (Table 2). Immunofluorescence revealed robust PMN infiltration based on MPO staining (Figure 1A). We also observed numerous citrullinated histone H3+ and MPO+ PMNs and rare lattices of extracellular DNA decorated with citrullinated histone H3 and MPO, demonstrating that PMNs early in the process of NET formation, as well as extracellular NETs, are present in the human lung during COVID-19 (Figure 1A; supplemental Figure 1, available on the Blood Web site). We measured plasma MPO-DNA complexes in 33 COVID-19 patients (n = 28 hospitalized, n = 5 convalescent) and 17 age- and sex-matched healthy donors to quantify plasma NET levels. We observed a significant increase in plasma NET levels in nonintubated COVID-19 patients, as well as in endotracheally intubated COVID-19 patients, compared with healthy donors and convalescent patients (P < .0001; Figure 1B). Plasma NET levels were significantly higher in COVID-19 nonsurvivors compared with COVID-19 survivors (P = .0004; Figure 1C).
Clinical course of autopsy patients who died from COVID-19 ARDS
| Case 1: Older patient with multiple preexisting medical conditions |
| This 64-year-old male of Hispanic decent with diabetes, end-stage renal disease on hemodialysis, heart failure, and hepatitis C on ledipasvir/sofosbuvir therapy developed fever after presenting with respiratory distress to the emergency room. SARS-CoV-2 PCR from a nasopharyngeal swab obtained prior to his demise was positive. He declined medical intervention, including intubation, and died within 5 hours of presentation. There was no clinical evidence of sepsis in this patient, premortem bacterial cultures were negative, and autopsy was conducted within 5 hours of his death. Neutrophil infiltration, but without immunofluorescence testing for NETs or platelets, of this patient’s autopsy lung sample has been published.4 |
| Case 2: Elderly with preexisting medical conditions and ARDS |
| This 73-year-old male with chronic obstructive pulmonary disease and diabetes developed ARDS with an arterial oxygen saturation of 50%. He was intubated and treated empirically with ceftriaxone, azithromycin, and doxycycline for community-acquired pneumonia with negative blood cultures. His chest radiograph showed diffuse patchy airspace opacification. SARS-CoV-2 PCR from a nasopharyngeal swab was positive. The patient required mechanical ventilation and experienced acute renal failure (creatinine increased from 2.4 to 4.1 mg/dL). His white blood cell count increased as did his absolute neutrophil count. He was lymphopenic. He remained afebrile with a temperature maximum of 37.8°C. He expired on hospital day 5 from COVID-19–related ARDS. |
| Case 3: Elderly with multiple preexisting medical conditions and cardiac arrest |
| This 71-year-old male with hypertension, hyperlipidemia, coronary artery disease, and diabetes had cough and fever for several days prior to a witnessed sudden cardiac arrest at home. His wife was a known SARS-CoV-2+ household exposure with minimal symptoms, similar to the patient’s initial presentation. Despite attempts at cardiopulmonary resuscitation by emergency medical personal in transit and in the emergency room, he expired. A SARS-CoV-2 nasopharyngeal swab was positive in the emergency room prior to his demise. |
| Case 1: Older patient with multiple preexisting medical conditions |
| This 64-year-old male of Hispanic decent with diabetes, end-stage renal disease on hemodialysis, heart failure, and hepatitis C on ledipasvir/sofosbuvir therapy developed fever after presenting with respiratory distress to the emergency room. SARS-CoV-2 PCR from a nasopharyngeal swab obtained prior to his demise was positive. He declined medical intervention, including intubation, and died within 5 hours of presentation. There was no clinical evidence of sepsis in this patient, premortem bacterial cultures were negative, and autopsy was conducted within 5 hours of his death. Neutrophil infiltration, but without immunofluorescence testing for NETs or platelets, of this patient’s autopsy lung sample has been published.4 |
| Case 2: Elderly with preexisting medical conditions and ARDS |
| This 73-year-old male with chronic obstructive pulmonary disease and diabetes developed ARDS with an arterial oxygen saturation of 50%. He was intubated and treated empirically with ceftriaxone, azithromycin, and doxycycline for community-acquired pneumonia with negative blood cultures. His chest radiograph showed diffuse patchy airspace opacification. SARS-CoV-2 PCR from a nasopharyngeal swab was positive. The patient required mechanical ventilation and experienced acute renal failure (creatinine increased from 2.4 to 4.1 mg/dL). His white blood cell count increased as did his absolute neutrophil count. He was lymphopenic. He remained afebrile with a temperature maximum of 37.8°C. He expired on hospital day 5 from COVID-19–related ARDS. |
| Case 3: Elderly with multiple preexisting medical conditions and cardiac arrest |
| This 71-year-old male with hypertension, hyperlipidemia, coronary artery disease, and diabetes had cough and fever for several days prior to a witnessed sudden cardiac arrest at home. His wife was a known SARS-CoV-2+ household exposure with minimal symptoms, similar to the patient’s initial presentation. Despite attempts at cardiopulmonary resuscitation by emergency medical personal in transit and in the emergency room, he expired. A SARS-CoV-2 nasopharyngeal swab was positive in the emergency room prior to his demise. |
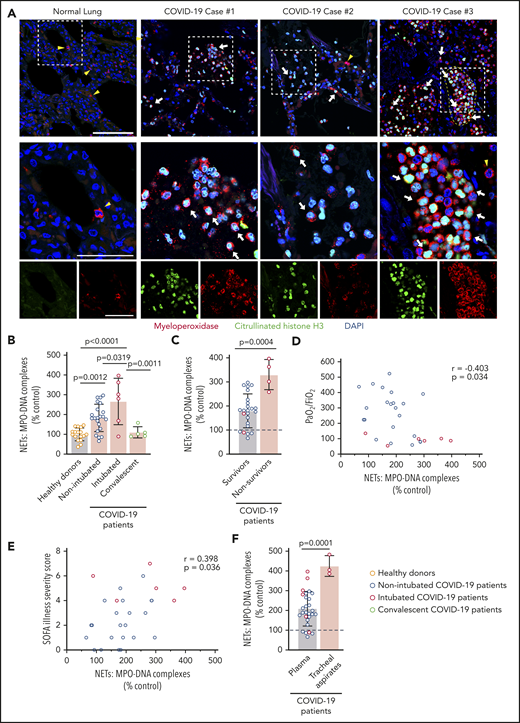
Increased plasma NETs correlate with increased COVID-19 severity. (A) We stained for neutrophils and NETs, using immunofluorescence, in lung tissue obtained at autopsy from COVID-19 patients (n = 3). Commercially available normal lung tissue was stained as a negative control. Neutrophils express MPO (red), and early-stage NET-forming neutrophils also express citrullinated histone H3 (green). DAPI serves as a nuclear DNA counterstain (blue). Cyan fluorescence represents the colocalization of citrullinated histone H3 with DNA. The yellow arrowheads point to neutrophils not making NETs, and the white arrows point to neutrophils making NETs. The dashed line highlights a thrombus in the microvasculature (top right panel). Scale bar, 100 μm. Higher-magnification images of the boxed areas in the top row (middle row) are shown along with single-channel images (bottom row); scale bars, 50 μm. (B-F) We used MPO-DNA ELISA to assess NETs in plasma and tracheal aspirate samples from patients in our COVID-19 prospective cohort and in age- and sex-matched healthy donors; each colored dot represents an individual participant. (B) We compared plasma NET levels across all groups: healthy adult donors (n = 17), adults hospitalized with COVID-19 but not intubated for ARDS (n = 22), adults intubated for COVID-19 ARDS (n = 6), and adults recovered from COVID-19 (n = 5). The y-axis depicts plasma NETs expressed as a percentage of healthy adult controls ± SD, arbitrarily set at 100%. (C) We compared plasma NET levels in 2 groups of COVID-19 hospitalized patients: survivors (n = 24) vs nonsurvivors (n = 4). The y-axis depicts plasma NETs expressed as a percentage of healthy adult donors ± SD, arbitrarily set at 100% (dashed line). (D) We correlated plasma NET levels with the Pao2/FiO2 ratio measure of respiratory failure for all hospitalized COVID-19 patients (n = 28). (E) We correlated plasma NET levels with the SOFA Illness Severity Scores for all hospitalized COVID-19 patients (n = 28). (F) We compared plasma NET levels in adult COVID-19 patients (n = 28) with NET levels quantified in the available tracheal aspirate samples of intubated COVID-19 patients (n = 3). The y-axis depicts NET levels expressed as a percentage of healthy adult donors ± SD, arbitrarily set at 100% (dashed line). One-way ANOVA with Tukey’s multiple-comparisons post hoc testing (B-C), Spearman’s rank-correlation test (D-E), Student t test (F).
Increased plasma NETs correlate with increased COVID-19 severity. (A) We stained for neutrophils and NETs, using immunofluorescence, in lung tissue obtained at autopsy from COVID-19 patients (n = 3). Commercially available normal lung tissue was stained as a negative control. Neutrophils express MPO (red), and early-stage NET-forming neutrophils also express citrullinated histone H3 (green). DAPI serves as a nuclear DNA counterstain (blue). Cyan fluorescence represents the colocalization of citrullinated histone H3 with DNA. The yellow arrowheads point to neutrophils not making NETs, and the white arrows point to neutrophils making NETs. The dashed line highlights a thrombus in the microvasculature (top right panel). Scale bar, 100 μm. Higher-magnification images of the boxed areas in the top row (middle row) are shown along with single-channel images (bottom row); scale bars, 50 μm. (B-F) We used MPO-DNA ELISA to assess NETs in plasma and tracheal aspirate samples from patients in our COVID-19 prospective cohort and in age- and sex-matched healthy donors; each colored dot represents an individual participant. (B) We compared plasma NET levels across all groups: healthy adult donors (n = 17), adults hospitalized with COVID-19 but not intubated for ARDS (n = 22), adults intubated for COVID-19 ARDS (n = 6), and adults recovered from COVID-19 (n = 5). The y-axis depicts plasma NETs expressed as a percentage of healthy adult controls ± SD, arbitrarily set at 100%. (C) We compared plasma NET levels in 2 groups of COVID-19 hospitalized patients: survivors (n = 24) vs nonsurvivors (n = 4). The y-axis depicts plasma NETs expressed as a percentage of healthy adult donors ± SD, arbitrarily set at 100% (dashed line). (D) We correlated plasma NET levels with the Pao2/FiO2 ratio measure of respiratory failure for all hospitalized COVID-19 patients (n = 28). (E) We correlated plasma NET levels with the SOFA Illness Severity Scores for all hospitalized COVID-19 patients (n = 28). (F) We compared plasma NET levels in adult COVID-19 patients (n = 28) with NET levels quantified in the available tracheal aspirate samples of intubated COVID-19 patients (n = 3). The y-axis depicts NET levels expressed as a percentage of healthy adult donors ± SD, arbitrarily set at 100% (dashed line). One-way ANOVA with Tukey’s multiple-comparisons post hoc testing (B-C), Spearman’s rank-correlation test (D-E), Student t test (F).
Next, we determined whether plasma NET levels in COVID-19 patients correlate with COVID-19 disease severity. We found that the ratio of Pao2/fraction of inspired oxygen (FiO2), a marker of severity of respiratory failure for which a lower score indicates increasing respiratory failure, varies inversely with plasma NETs (P = .0340; Figure 1D). Plasma NETs also correlate directly with the Sequential Organ Failure Assessment (SOFA) score, a clinical illness severity score (P = .0360; Figure 1E). NET levels were also significantly higher in tracheal aspirate fluid than in plasma samples in COVID-19 patients (P = .0001; Figure 1F). Following recovery from COVID-19, plasma NET levels decreased to levels similar to those of healthy adults, suggesting that NET formation normalizes during convalescence after COVID-19 (Figure 1B; supplemental Figure 2).
COVID-19 PMNs demonstrate increased activation and NETs at baseline and fail to respond to PMA with increased NET formation
We next assessed neutrophil granulation, using flow cytometry, in PMNs isolated from COVID-19 patients or healthy adult donors. We detected a significant decrease in PMN granularity in COVID-19 PMNs compared with those from healthy adults, suggesting neutrophil activation prior to isolation (P < .0001; Figure 2A). We then assessed NET formation in vitro in these PMNs. We demonstrated markedly elevated baseline NET levels in PMNs isolated from COVID-19 patients, whereas PMNs isolated from healthy adults remained unaltered under basal conditions (P = .0012; Figure 2B-C). We also demonstrated that PMA, a nonphysiologic agonist, induced NETs in vitro by PMNs isolated from healthy adults, as previously reported,23 but failed to further increase NET formation over the already high baseline in COVID-19 PMNs (Figure 2B-C). Although PMNs do not express Toll-like receptor 3 (TLR3),24,25 we also found that poly(I:C), a damage-associated molecular pattern that typically signals through TLR3, induces NETs in PMNs isolated from COVID-19 adults and healthy donors (supplemental Figure 3).
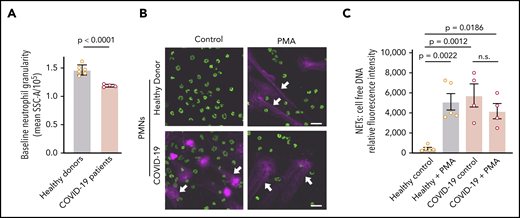
COVID-19 PMNs demonstrate increased activation and NETs at baseline and fail to respond to PMA with increased NET formation. (A) We assessed granularity using flow cytometry in PMNs isolated from healthy donors (n = 6) and COVID-19 patients (n = 4) as a measure of baseline PMN activation. (B-C) We assessed NETs in PMNs isolated from healthy donors (n = 5) and COVID-19 patients (n = 4), with and without PMA stimulation (20 nM; 2 hours), using confocal microscopy and cell-free DNA quantitation. (B) Representative images show NETs (magenta; white arrows) and nuclear DNA (green). Scale bars, 20 μm. (C) We quantified NETs using a fluorescence-based cell-free DNA assay as described in Patients and methods. The y-axis depicts NETs shown as relative cell-free DNA fluorescence ± SEM. Student t test (A), 1-way ANOVA with Tukey’s multiple-comparisons post hoc test (C). n.s., not significant.
COVID-19 PMNs demonstrate increased activation and NETs at baseline and fail to respond to PMA with increased NET formation. (A) We assessed granularity using flow cytometry in PMNs isolated from healthy donors (n = 6) and COVID-19 patients (n = 4) as a measure of baseline PMN activation. (B-C) We assessed NETs in PMNs isolated from healthy donors (n = 5) and COVID-19 patients (n = 4), with and without PMA stimulation (20 nM; 2 hours), using confocal microscopy and cell-free DNA quantitation. (B) Representative images show NETs (magenta; white arrows) and nuclear DNA (green). Scale bars, 20 μm. (C) We quantified NETs using a fluorescence-based cell-free DNA assay as described in Patients and methods. The y-axis depicts NETs shown as relative cell-free DNA fluorescence ± SEM. Student t test (A), 1-way ANOVA with Tukey’s multiple-comparisons post hoc test (C). n.s., not significant.
NETs associate with microthrombi formation and platelet deposition in COVID-19 patients
In additional studies of the autopsy lung samples (Table 2), many of the neutrophils undergoing early-stage NETosis localized in structures consistent with blood vessels (Figures 1A, 3A). Using immunofluorescence, we demonstrated colocalization of citrullinated histone H3+ neutrophils, likely undergoing NETosis, with platelets (staining with PF4) in structures consistent with blood vessels in the lung (Figure 3A), suggesting that NETs interact with platelets in COVID-19, a mechanism that may contribute to thrombosis in COVID-19 ARDS and perhaps throughout the body. Although we did not see the many thrombotic complications of SARS-CoV-2 infection in our cohort that other investigators have described, this most likely represents clinical testing limitations at our institution that preclude imaging studies in SARS-CoV-2 patients. However, we detected significantly higher levels of circulating platelet-neutrophil aggregates in COVID-19 patients compared with healthy adults (P = .006; Figure 3B; supplemental Figure 4). Soluble markers of thrombosis (eg, plasma D-dimer and VWF antigen levels) were also significantly elevated in COVID-19 patients (P < .0001 and P = .0077, respectively; Figure 3C-D), although they did not correlate directly with plasma NET levels in our cohort. Finally, we observed significantly elevated levels of soluble platelet-derived factors that trigger NETosis, including PF4 and RANTES, in COVID-19 patients (both P < .0001; Figure 3E-F). These data suggest that, in COVID-19 patients, the interplay of NETs with platelets may contribute to a thrombo-inflammatory cascade leading to the COVID-19 hypercoagulability and thrombosis that are observed clinically.
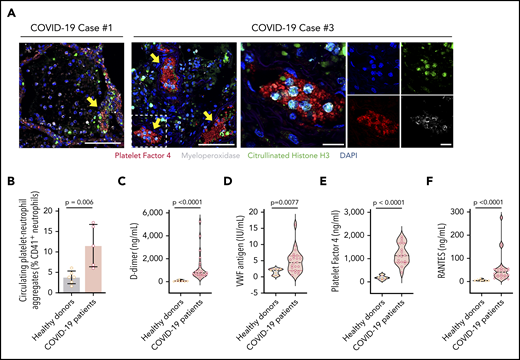
NETs associate with microthrombi formation and platelet deposition in COVID-19 patients. (A) We stained for NETs and platelets using immunofluorescence in COVID-19 lung tissue obtained from autopsy cases #1 and #3. Early-stage NET-forming neutrophils (MPO, white) express citrullinated histone H3 (green). Microthrombi stained for early-stage NET-forming neutrophils and platelets (PF4; red). DAPI served as a nuclear DNA counterstain (blue). Scale bars, 100 μm (low-magnification, 2 leftmost images). For case #3, higher-magnification images are presented along with single-channel images. Scale bars, 20 μm. Yellow arrows point to thrombi with early-stage NET-forming neutrophils. Colocalized PF4 and early stage NET-forming neutrophils were not present in the analyzed lung sample from case #2. (B) We used flow cytometry to quantify platelet-neutrophil aggregates in whole blood from COVID-19 patients (n = 5) and healthy donors (n = 6). We used ELISA to quantify plasma levels of D-dimer (C), VWF antigen (D), PF4 (E), and RANTES (F) in available plasma from 18 to 22 COVID-19 patients and 5 to 7 healthy donors. Student t test (B), Mann-Whitney or Student t test, depending on the normality of distribution (C-F).
NETs associate with microthrombi formation and platelet deposition in COVID-19 patients. (A) We stained for NETs and platelets using immunofluorescence in COVID-19 lung tissue obtained from autopsy cases #1 and #3. Early-stage NET-forming neutrophils (MPO, white) express citrullinated histone H3 (green). Microthrombi stained for early-stage NET-forming neutrophils and platelets (PF4; red). DAPI served as a nuclear DNA counterstain (blue). Scale bars, 100 μm (low-magnification, 2 leftmost images). For case #3, higher-magnification images are presented along with single-channel images. Scale bars, 20 μm. Yellow arrows point to thrombi with early-stage NET-forming neutrophils. Colocalized PF4 and early stage NET-forming neutrophils were not present in the analyzed lung sample from case #2. (B) We used flow cytometry to quantify platelet-neutrophil aggregates in whole blood from COVID-19 patients (n = 5) and healthy donors (n = 6). We used ELISA to quantify plasma levels of D-dimer (C), VWF antigen (D), PF4 (E), and RANTES (F) in available plasma from 18 to 22 COVID-19 patients and 5 to 7 healthy donors. Student t test (B), Mann-Whitney or Student t test, depending on the normality of distribution (C-F).
nNIF blocks NETs induced by soluble factors in plasma from COVID-19 patients
We next demonstrated that incubation of PMNs, isolated from healthy adults, with plasma from COVID-19 patients induces robust NET formation, whereas plasma from healthy adults does not (Figure 4). Our analysis of COVID-19 plasma showed significantly elevated levels of 2 additional NET-inducing cytokines compared with healthy adults: IL-6 (53.49 ± 10.91 vs 0.3626 ± 0.2055 pg/mL; P = .0017) and IL-8 (17.52 ± 12.26 vs 4.236 ± 1.280 pg/mL; P = .0058).26 Other investigators have reported elevated plasma IL-6 and IL-8 levels in COVID-19 patients.27
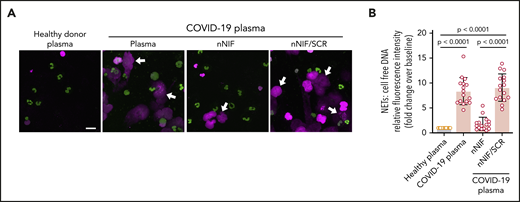
nNIF blocks NETs induced by soluble factors in plasma from COVID-19 patients. (A) We used confocal microscopy to assess NET formation qualitatively. We isolated PMNs from healthy adults (n = 4) and used those for experiments testing 16 different available COVID-19 patient plasma samples for NET inductive capacity. We used plasma from age- and sex-matched healthy donors as a control (n = 10). We incubated PMNs with control plasma or COVID-19 patient plasma and assessed for NET formation after a 2-hour incubation. PMNs were preincubated for 1 hour with nNIF (1 nM) or its inactive scrambled peptide control (SCR; 1 nM). Representative images from each experimental arm show NETs (magenta; white arrows) and nuclear DNA (green). Scale bar, 20 μm. (B) We used a high throughput cell-free DNA fluorescence assay to quantify NET formation in the PMNs treated in (A). The y-axis depicts NETs measured as fold change over baseline relative fluorescence units ± SEM. The PMNs treated with control healthy donor plasma serve as the baseline PMN NET level, arbitrarily set at 1. The P values were determined using 1-way ANOVA with Tukey’s multiple-comparisons post hoc testing.
nNIF blocks NETs induced by soluble factors in plasma from COVID-19 patients. (A) We used confocal microscopy to assess NET formation qualitatively. We isolated PMNs from healthy adults (n = 4) and used those for experiments testing 16 different available COVID-19 patient plasma samples for NET inductive capacity. We used plasma from age- and sex-matched healthy donors as a control (n = 10). We incubated PMNs with control plasma or COVID-19 patient plasma and assessed for NET formation after a 2-hour incubation. PMNs were preincubated for 1 hour with nNIF (1 nM) or its inactive scrambled peptide control (SCR; 1 nM). Representative images from each experimental arm show NETs (magenta; white arrows) and nuclear DNA (green). Scale bar, 20 μm. (B) We used a high throughput cell-free DNA fluorescence assay to quantify NET formation in the PMNs treated in (A). The y-axis depicts NETs measured as fold change over baseline relative fluorescence units ± SEM. The PMNs treated with control healthy donor plasma serve as the baseline PMN NET level, arbitrarily set at 1. The P values were determined using 1-way ANOVA with Tukey’s multiple-comparisons post hoc testing.
Given that an inhibitor of NETosis might reduce dysregulated immunothrombosis, we next assessed a novel approach to inhibit NET formation. Previously, our laboratory discovered a class of endogenous NET-inhibitory peptides circulating in human umbilical cord blood.18,19 These NET-inhibitory peptides are cleavage fragments of the C terminus of α-1-antitrypsin, a serine protease inhibitor. We found that nNIF, a NET-inhibitory peptide that we synthesized based on the endogenous sequence, significantly decreased NETs induced in vitro by COVID-19 plasma, whereas an inactive scrambled peptide control did not (Figure 4).
Discussion
In this prospective cohort study, we demonstrate a robust correlation between NETs and the severity of respiratory illness in COVID-19. We measured the highest levels of circulating NETs in COVID-19 patients with endotracheal intubation and report, for the first time, the infiltration of platelet colocalization with citrullinated histone H3+ neutrophils likely undergoing NETosis in the pulmonary microthrombi of patients who died from COVID-19. Our combined data suggest that NETs contribute to COVID-19–related lung injury and microthrombi, similar to reports in other non–COVID-19 viral and autoimmune NET-related ARDS.12,28 Along with a recent position paper4 and case series,29 our study provides additional clinical evidence of the potential importance of NETs in the pathogenesis of COVID-19–related ARDS and coagulopathy.
Although we enrolled a limited number of COVID-19 patients and lack long-term follow-up for ischemic events, our study represents the first prospective cohort assembled to examine the effects of NETosis in COVID-19. In contrast to previous reports,29 our study analyzes plasma samples collected prospectively from COVID-19 patients at the hospital bedside as opposed to discarded serum samples. Therefore, we can successfully correlate plasma NET levels with clinical outcomes in our study population. Evidence of increased NETs measured in the plasma samples from our intubated vs nonintubated patient cohort suggests that plasma NETs may serve as a prognostic indicator and predict survival in COVID-19; however, further studies will be required in larger COVID-19 patient cohorts.
NETs were first described in 2004; they can contribute to immunothrombosis, defined as a physiologic thrombosis that enhances the innate immune response against pathogenic insults.30,31 Representing a primitive defense against microbial invaders, PMNs release NETs to physically capture and eliminate infections.11 First recognized for their role in only bacterial clearance, NETs are now firmly established as part of the innate immune defense against a wide repertoire of RNA-stranded viral pathogens, including respiratory syncytial virus, influenza, dengue, and even HIV.6,18 Dysregulated NETosis triggered by respiratory viruses can lead to excessive acute lung injury,12,32,33 similar to the lung pathology that we observe in COVID-19 ARDS.
Release of NETs by PMNs can be stimulated by many physiologic and nonphysiologic agonists, such as lipopolysaccharide, live microbes or viruses, poly(I:C), and chemical agonists (eg, PMA). We chose only PMA and poly(I:C) to test NET inductive capacity for PMNs isolated from COVID-19 patients, and we recognize that our study does not represent a comprehensive evaluation of NET induction by various agonists in COVID-19 patients. Furthermore, our finding of NET induction by poly(I:C) was unexpected, because PMNs do not express TLR3.25 However, other receptors for RNA viral mimetic damage-associated molecular patterns are expressed by the PMNs, RIG1, MDA-5, and TLR8.24 We also observed a 50-fold increase in NET release in COVID-19 PMNs at baseline compared with healthy controls, and we triggered a robust NET induction in healthy PMNs exposed to COVID-19 patient plasma. Our findings support that the rapid release of NETs in COVID-19 contributes to their maladaptive potential and that NETs may be responsible for propagation of thrombi in the vasculature, leading to ARDS and multiorgan failure similar to other systemic inflammatory syndromes.11,34,35
The potential involvement of NETs in COVID-19 is further supported by the well-described recruitment of neutrophils in the progression of SARS-CoV-2 pulmonary infection. Neutrophils, the effector cell for NET production, increase dramatically with COVID-19 severity and ARDS.36,37 In addition, NET cytokine signatures have been demonstrated following SARS-CoV-2 infection in patient lungs (eg, IL-1, tumor necrosis factor)38 and serum (eg, IL-6).27 As described above, we identified microvascular thrombi with neutrophils releasing NETs mixed with platelets in the postmortem examination of lungs of COVID-19 patients, and this was most pronounced in the 2 patients with rapid disease progression and sudden death. Further supporting the role of NETs in SARS-CoV-2 pulmonary pathophysiology, here we offer an initial description of high levels of NETs in tracheal aspirates from intubated COVID-19 patients. We also preliminarily observe, for the first time, that NET levels may return to normal in convalescent patients (Figure 1B; supplemental Figure 2). When coupled with an inverse correlation with oxygenation (Pao2/FiO2) and a direct correlation with SOFA scores, our findings suggest that NETs released from recruited neutrophils to fight SARS-CoV-2 may do more harm than good in the lungs of patients with COVID-19.
Similar to NETs, platelets play a role in the intravascular immune response and, if unchecked, contribute to inflammatory sequelae in sepsis and COVID-19.3,39 Platelets detect foreign pathogens through pattern recognition receptors (eg, TLRs),40 including the detection of viruses, and they coordinate with PMNs to release NETs through chemokine and coagulation factor signaling (eg, PF4, HMGB1, VWF).41 In our cohort, we found elevated levels of PF4 and platelet-neutrophil aggregates in COVID-19 patients. PF4, released from platelets, binds to NETs, making them compact and DNase resistant,42 and PF4 can also be released from platelets trapped inside thrombogenic NETs. This PF4/NET positive-feedback loop initiates and sustains a coagulative NET cascade that might explain, in part, the high levels of PF4 and NETs observed in COVID-19 patients.
COVID-19 can present clinically anywhere along the thrombotic spectrum, ranging from otherwise asymptomatic patients with ischemic cerebral events and myocardial infarctions to ICU patients with disseminated intravascular coagulopathy, stroke, venous embolism, and multiorgan failure.43-45 In an effort to protect imaging technicians during this pandemic, current clinical guidelines at our institution preclude screening for thrombosis in COVID-19. Therefore, we cannot report the incidence of thrombotic complications in our cohort of COVID-19 patients. However, thromboembolic events occur frequently in COVID-1946 ; therefore, the International Society of Thrombosis and Haemostasis recommends prophylactic low molecular weight heparin for all hospitalized patients with COVID-19.47,48 Yet many patients on prophylactic heparin remain at risk for thrombotic complications. We hypothesize that, in COVID-19, NETs and platelets increase the risk for thrombosis with potential pathologic sequelae that may contribute to heparin resistance and require higher treatment doses. Our data support, but do not prove, this hypothesis; further studies are clearly warranted.
We report high levels of markers associated with thrombosis, including VWF, D-dimers, and PF4, but acknowledge that plasma levels of these thrombotic markers do not correlate with plasma NET levels in our COVID-19 cohort. This may result from our small sample size. The complicated role of NETs in thrombosis may also contribute in several ways to the lack of correlation with VWF and D-dimers. First, we measured VWF antigen levels and not activity, and VWF antigen levels do not reflect the ability of VWF to interact with platelets and PMNs.49 Second, VWF is released by activated and damaged endothelium, which likely occurs early in COVID-19. Our plasma VWF levels were measured later in the disease process during hospitalization, and the very high levels that we observed may be confounded by ongoing inflammation. Interestingly, NETs themselves will induce endothelial cell damage, resulting in ongoing VWF release.50 Third, plasma D-dimers originate from active fibrinolysis, and NET-rich clots are protected from fibrinolytic breakdown by tissue plasminogen activator.7 The lack of correlation between NETs and D-dimers may reflect inhibited thrombus degradation in patients with COVID-19.
Considered together, we propose that the clinical manifestations of severe COVID-19 may be explained by NETs and dysregulated immunothrombosis in response to SARS-CoV-2. Although we and other investigators have shown that microthrombi with fibrin deposits are present in the lungs of patients with COVID-19 ARDS,4,51,52 we report that citrullinated histone H3+ neutrophils, likely undergoing NETosis, colocalize with platelet-derived thrombotic factors in pulmonary microthrombi. Neutrophils from COVID-19 patients seem especially prone to robust and excessive NET release. Moreover, disease severity and survival parallel increasing markers of NET formation. Our findings suggest that NET-related immunothrombosis may offer insight into the puzzling observation of overwhelming inflammation with cytokine storm and widespread microvascular and macrovascular thrombosis in COVID-19 patients.53,54 If this is the case, then the disintegration or inhibition of NETs represent enticing targets for clinical intervention in this COVID-19 pandemic.
Clinical studies continue to be conceptualized to target dysregulated NET formation in COVID-19 patients, including trials of DNase (NCT04359654) and dipyridamole (NCT04391179).4 Here, we present evidence that synthesized nNIF, an inhibitor of NETosis discovered in human umbilical cord blood,18 effectively blocks NETs induced by COVID-19 plasma in PMNs from healthy adults. Previous testing of nNIF in preclinical models of sepsis demonstrates decreased severity and improved survival with an accompanying blockade of NETs,18 a finding that could be clinically relevant to COVID-19 given the extremely high rate of patients who die of sepsis. Although our study correlates NETs with clinical progression and respiratory failure, further studies will need to prove that targeting NETs ameliorates COVID-19 sepsis and ARDS. Currently, nNIF and related NET-inhibitory peptides continue to be evaluated in the preclinical setting for their therapeutic potential in COVID-19 and other diseases associated with immunothrombosis. Effectively blocking NETosis in COVID-19 patients with naturally occurring peptides like nNIF may decrease thromboembolic events and improve clinical outcomes for COVID-19. Given the pathogenic role of dysregulated NET formation in non–COVID-19 diseases,11-13,55 developing medicines to inhibit NETs may have broader clinical application to other inflammatory and thrombotic conditions of the lung.
Data sharing requests should be sent to Christian Con Yost (christian.yost@u2m2.utah.edu).
The online version of this article contains a data supplement.
The publication costs of this article were defrayed in part by page charge payment. Therefore, and solely to indicate this fact, this article is hereby marked “advertisement” in accordance with 18 USC section 1734.
Acknowledgments
The authors acknowledge Diana Lim for expertise in figure preparation; Antoinette Blair, Macy Barrios, Amber Plante, Jordan Greer, Amy DeNardo, Amanda Bailey, and Lindsey Waddoups for assistance with participant recruitment; and the “NETwork to Target Neutrophils in COVID-19” for meaningful discussions.
This work was supported by grants from the National Institutes of Health (NIH)/National Institute of Children's Health and Development (R01 HD093826 [C.C.Y.]), NIH/National Cancer Institute (5P30CA045508-30 [M.E.]), NIH/National Heart, Lung, and Blood Institute (R01 HL135265 [A.C.P.]; R35 HL145237, R01 HL142804NIA, and R61HL141783 [A.S.W.]), and NIH/National Institute on Aging (K01 AG059892 [R.A.C.]); the University of Utah Triple I Program (E.A.M.); Fonds voor Wetenschappelijk Onderzoek Vlaanderen FWO 12U7818N (F.D.); Animal Cancer Foundation (J.D.S.); Soccer For Hope Foundation (J.D.S.); Closer To Cure Foundation (J.D.S.); and PEEL Therapeutics, Inc (C.C.Y.). This work was also supported, in part, by Merit Review Award I01 CX001696 (M.T.R.) from the US Department of Veterans Affairs Clinical Sciences R&D Service. M.E., X.-Y.H., and D.N. were supported by the William C. and Joyce C. O’Neill Charitable Trust and the Linartz-Meier Family Foundation. This article is the result of work supported with resources and the use of facilities at the George E. Wahlen VA Medical Center.
The contents do not represent the views of the US Department of Veterans Affairs or the US Government.
Authorship
Contribution: E.A.M. recruited and obtained consent from study participants, performed, directed, and interpreted experiments, and wrote sections of the manuscript; X.-Y.H. and D.N. performed autopsy immunofluorescence staining and wrote sections of the manuscript; S.P.S., M.M., A.B.-S., A.C.B., and M.L. performed autopsies on COVID-19 patients and composed clinical vignettes for each autopsy case; F.D., M.J.C., and R.A.C. performed experiments, analyzed data, and wrote sections of the manuscript; E.S.H. recruited and obtained consent from study participants and analyzed study participant clinical data; B.K.M., I.P., A.C.P., and E.J.B. performed experiments and analyzed data; A.F.C., A.I., L.M.A., A.S.W., M.T.R., and M.E. analyzed data and contributed to the writing of the manuscript; and J.D.S. and C.C.Y. provided overall direction for the project, reviewed and analyzed data from all experiments, wrote sections of the manuscript, and edited the manuscript.
Conflict-of-interest disclosure: C.C.Y. has received grant funding from PEEL Therapeutics, Inc. during the conduct of this study. In addition, C.C.Y. authored a United States patent (patent no. 10 232023 B2) held by the University of Utah for the use of NET-inhibitory peptides for the “treatment of and prophylaxis against inflammatory disorders,” for which PEEL Therapeutics, Inc. holds the exclusive license. A.I. and L.M.A. are consultants and stock option holders of PEEL Therapeutics, Inc., and A.F.C. and J.D.S. are employees and stock option holders of PEEL Therapeutics, Inc. The remaining authors declare no competing financial interests.
Correspondence: Christian Con Yost, Department of Pediatrics, University of Utah School of Medicine, Eccles Institute of Human Genetics, Room 4220A, 15 N 2030 East, Salt Lake City, UT 84112; e-mail: christian.yost@u2m2.utah.edu.
