


Abstract
The mature red blood cell (RBC) lacks a nucleus and organelles characteristic of most cells, but it is elegantly structured to perform the essential function of delivering oxygen and removing carbon dioxide from all other cells while enduring the shear stress imposed by navigating small vessels and sinusoids. Over the past several decades, the efforts of biochemists, cell and molecular biologists, and hematologists have provided an appreciation of the complexity of RBC membrane structure, while studies of the RBC membrane disorders have offered valuable insights into structure–function relationships. Within the last decade, advances in genetic testing and its increased availability have made it possible to substantially build upon this foundational knowledge. Although disorders of the RBC membrane due to altered structural organization or altered transport function are heterogeneous, they often present with common clinical findings of hemolytic anemia. However, they may require substantially different management depending on the underlying pathophysiology. Accurate diagnosis is essential to avoid emergence of complications or inappropriate interventions. We propose an algorithm for laboratory evaluation of patients presenting with symptoms and signs of hemolytic anemia with a focus on RBC membrane disorders. Here, we review the genotypic and phenotypic variability of the RBC membrane disorders in order to raise the index of suspicion and highlight the need for correct and timely diagnosis.
Introduction
The mammalian erythrocyte has evolved as a highly differentiated cell to deliver oxygen throughout the circulation, with a structure optimized to maintain survival under continuous shear stress. The lipid bilayer of the cell membrane is lined on its cytoplasmic surface by the erythrocyte cytoskeleton, and together they compose the engineering marvel we call the red blood cell (RBC) membrane (Figure 1).1,2 The scaffolding network of the cytoskeleton has a design of triangles weaved in a hexagonal network. Two α- and β-spectrin heterodimers are organized in an antiparallel fashion into a tetramer making up each side of the triangle. Each heterodimer is built from a series of 3-helix bundles (spectrin repeats) able to coil and uncoil, providing elasticity. The junctional complex, composed of an F-actin protofilament with its capping proteins adducin and tropomodulin, and protein 4.1R that enables the actin-spectrin association,3 is positioned at each corner of the triangle. The length of the actin filaments is maintained fairly stable at 12 to 14 actin monomers per oligomer,1,4 indicating the existence of strict mechanisms that control the filament length.5-7 Work by multiple research teams over decades has determined the stoichiometry of the various proteins in the cytoskeleton.1,8-10 Based on recent proteomic quantification of highly purified populations of reticulocytes and mature RBCs, actin monomers are calculated to be >1 million copies per cell and β-spectrin ∼0.5 million copies per cell.10 Consequently, assuming 12 actin monomers per protofilament, the quasihexagonal lattice of the RBC cytoskeleton is estimated to have ∼80 000 junctional complexes linked with ∼250 000 spectrin tetramers.11 Band 3, at ∼1.2 million copies per cell,8 arranges in tetramers and dimers and creates >300 000 vertical linkages between the cytoskeleton and the lipid bilayer. PIEZO1, the mechanosensitive cation channel, is present only at a few hundred copies per cell10 but functions as a major determinant of the RBC hydration status. The extensive horizontal protein–protein interactions in the cytoskeleton with perpendicular transmembrane channels, which serve as vertical linkages between the cytoskeleton and the cell membrane, maintain the cell’s biconcave disk shape, securing an increased surface area-to-volume ratio and allow the cell to reversibly deform while traversing capillaries with cross section as small as one-third the RBC diameter or passing through the interendothelial slits of the splenic red-pulp sinusoids.2
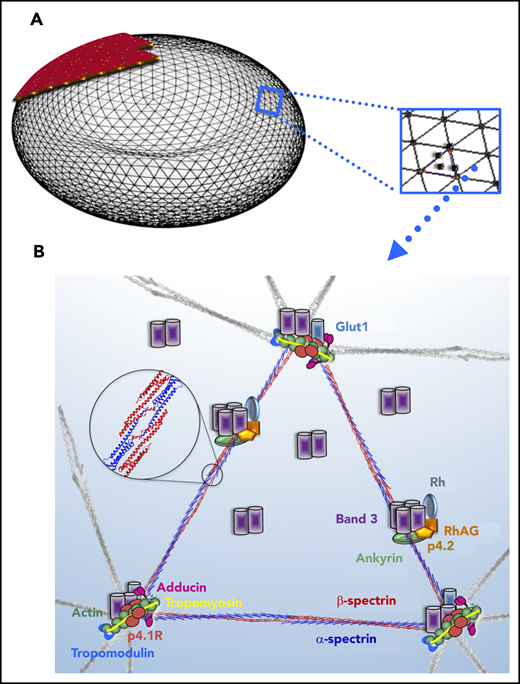
RBC membrane model depicting the structural proteins that, when abnormal, cause RBC membrane disorders. (A) Model of the RBC cytoskeleton quasihexagonal lattice forming a biconcave disc shape, supporting the lipid bilayer. An area of the cytoskeleton surface is shown “magnified” to demonstrate the arrangement of proteins within the hexagonal structure, focusing particularly on the proteins that, when defective, cause RBC membrane disorders. Illustration by Anastasios Manganaris (created using “blender” software, version 2.8). (B) α- and β-spectrin heterodimers associate head to head, as shown in the magnified circle, to form the spectrin tetramers that make the sides of each triangular unit in the hexagon. Each dimer head is composed of the N-terminal region of α-spectrin and the C-terminal region of β-spectrin. The junctional complex, at the corner of each triangle, is formed by an actin oligomer, with a length guided by tropomyosin, and capped by adducin and tropomodulin. Protein 4.1R enables the actin-spectrin association. The transmembrane protein complexes containing the integral membrane proteins band 3 and Rh-associated glycoprotein (RhAG) and the peripheral membrane proteins ankyrin and band 4.2 provide “vertical” linkages between the cytoskeleton and the lipid bilayer.
RBC membrane model depicting the structural proteins that, when abnormal, cause RBC membrane disorders. (A) Model of the RBC cytoskeleton quasihexagonal lattice forming a biconcave disc shape, supporting the lipid bilayer. An area of the cytoskeleton surface is shown “magnified” to demonstrate the arrangement of proteins within the hexagonal structure, focusing particularly on the proteins that, when defective, cause RBC membrane disorders. Illustration by Anastasios Manganaris (created using “blender” software, version 2.8). (B) α- and β-spectrin heterodimers associate head to head, as shown in the magnified circle, to form the spectrin tetramers that make the sides of each triangular unit in the hexagon. Each dimer head is composed of the N-terminal region of α-spectrin and the C-terminal region of β-spectrin. The junctional complex, at the corner of each triangle, is formed by an actin oligomer, with a length guided by tropomyosin, and capped by adducin and tropomodulin. Protein 4.1R enables the actin-spectrin association. The transmembrane protein complexes containing the integral membrane proteins band 3 and Rh-associated glycoprotein (RhAG) and the peripheral membrane proteins ankyrin and band 4.2 provide “vertical” linkages between the cytoskeleton and the lipid bilayer.
Patients with RBC cytoskeleton or hydration disorders due to altered structural organization or abnormal transporter function, respectively, present with the common clinical findings of hemolytic anemia, including pallor, jaundice, fatigue, splenomegaly, gallstones, and cholecystitis. However, they may require different management approaches depending on the cause of the disease. Therefore, accurate diagnosis is critical. A proposed algorithm for laboratory evaluation of a patient presenting with symptoms and signs of hemolysis with or without anemia with a focus on RBC membrane disorders is shown in Figure 2.
![Evaluation workflow of patients with hemolytic anemia. (A) Proposed algorithm for the laboratory evaluation of a patient presenting with symptoms and signs of hemolysis. Of note, anemia may be well compensated, as in many cases of mild HS and most cases of PIEZO1-associated HX. Evaluation for autoimmune or, especially in an infant, alloimmune hemolytic anemia with direct and indirect antiglobulin test (DAT and IAT) is the first priority, since such a diagnosis typically requires immediate action. Consideration of the possibility of microangiopathic hemolytic anemia (MAHA) and paroxysmal nocturnal hemoglobinuria (PNH) may also be necessary. Blood smear review of the patient and parents, with attention to the RBC indices, including MCV, MCHC, and red blood cell distribution width (RDW), along with hemolytic markers (bilirubin, lactate dehydrogenase, and haptoglobin [the last one reliable after 6 months of life, since earlier, it may be low due to decreased production by the infant’s liver]), ferritin, and transferrin saturation to consider iron-loading inefficient erythropoiesis, can guide the differential diagnosis. Flow cytometry with eosin-5′-maleimide (EMA) binding of band 3 and Rh-related proteins is a rapid screening test for RBC membrane disorders that are characterized by membrane loss.96,97 Osmotic fragility is increased in HS and often decreased in HX. Osmotic gradient ektacytometry, which evaluates the deformability of RBCs as they are subjected to constant shear stress in a medium of increasing osmolality in a laser diffraction viscometer, is the reference technique for differential diagnosis of erythrocyte membrane and hydration disorders when a recent transfusion does not interfere with phenotypic evaluation of the patient.98-100 When the patient is recently or chronically transfused, as it is typically the case in young children with congenital severe hemolytic anemia, options for phenotypic evaluation are limited. In such cases, genetic evaluation with clinically available next-generation sequencing panels may provide an accurate diagnosis necessary for appropriate management decisions.12,101,102 aHUS, atypical hemolytic uremic syndrome; HUS, hemolytic uremic syndrome; TTP, thrombotic thrombocytopenic purpura. (B) Osmotic gradient ektacytometry. The ektacytometry curve is determined by the RBC structural features.99 The points indicated in red are the following: Omin corresponds to the value of the hypotonic osmolality at which 50% of the cells hemolyze in an osmotic fragility assay and provides information on the initial surface to volume ratio of the RBCs. A shift to the right reflects a decrease in the surface area to volume ratio (ie, increased osmotic fragility). EImax corresponds to the maximum deformability of the RBC, and its value is affected by the cytoskeleton mechanics. The hypertonic descending part of the curve is represented by Ohyp, the osmolality value at which the cells’ average maximum diameter is half of EImax. The value of Ohyp correlates with the initial intracellular viscosity of the cell sample. A shift to the left reflects increased intracellular viscosity of the erythrocyte caused by increased intracellular concentration of hemoglobin, typically due to dehydration of the cell; a shift to the right may represent an overhydration state of the cell in overhydrated stomatocytosis or, most commonly, a decreased intracellular concentration of hemoglobin, such as in iron deficiency. (C) Typical osmotic gradient ektacytometry curves for various RBC membrane disorders (in red) in comparison with a normal control curve run concurrently (in green). (i) HS characterized by increased Omin and decreased EImax. (ii) HE/HPP characterized by decreased EImax and a trapezoid shape of the curve. (iii) HX with decreased Omin and decreased Ohyp. (iv) SAO with severely decreased deformability and decreased Omin.](https://ash.silverchair-cdn.com/ash/content_public/journal/blood/136/11/10.1182_blood.2019000946/1/m_bloodbld2019000946cf2.png?Expires=1769176587&Signature=umdwPonlFmAns5lSlA9LuhZt7dKHY-cE83ET5e88EOdu8MOE8r8~xb6ul6FchEyDMDhTql0ghCZUfAjeWLfpzJcGFGFhu0Q4SDlzS4V2UcxFamSh3lUUw9Yxf0Keq45EyApMZKVRzl-Xb9aAj0e1XJt6C~kAIjDjmWWreoQBc--Lbpr0PgysqrMS0qMgyZEF0Oho4skuXSOIJyTw2pSnRNt~lypW5CSlVfbkA3vWc0u-FWlUJEHyj4ZA3CUpswW5cFvUcQqjHs~s1-tNqYpRUULdUDUrenhOp-Cl0CXwV4NgezBg-4eAmlT~tEi8eSJ7IBk366r0nG5rdxOiEcq-Lg__&Key-Pair-Id=APKAIE5G5CRDK6RD3PGA)
Evaluation workflow of patients with hemolytic anemia. (A) Proposed algorithm for the laboratory evaluation of a patient presenting with symptoms and signs of hemolysis. Of note, anemia may be well compensated, as in many cases of mild HS and most cases of PIEZO1-associated HX. Evaluation for autoimmune or, especially in an infant, alloimmune hemolytic anemia with direct and indirect antiglobulin test (DAT and IAT) is the first priority, since such a diagnosis typically requires immediate action. Consideration of the possibility of microangiopathic hemolytic anemia (MAHA) and paroxysmal nocturnal hemoglobinuria (PNH) may also be necessary. Blood smear review of the patient and parents, with attention to the RBC indices, including MCV, MCHC, and red blood cell distribution width (RDW), along with hemolytic markers (bilirubin, lactate dehydrogenase, and haptoglobin [the last one reliable after 6 months of life, since earlier, it may be low due to decreased production by the infant’s liver]), ferritin, and transferrin saturation to consider iron-loading inefficient erythropoiesis, can guide the differential diagnosis. Flow cytometry with eosin-5′-maleimide (EMA) binding of band 3 and Rh-related proteins is a rapid screening test for RBC membrane disorders that are characterized by membrane loss.96,97 Osmotic fragility is increased in HS and often decreased in HX. Osmotic gradient ektacytometry, which evaluates the deformability of RBCs as they are subjected to constant shear stress in a medium of increasing osmolality in a laser diffraction viscometer, is the reference technique for differential diagnosis of erythrocyte membrane and hydration disorders when a recent transfusion does not interfere with phenotypic evaluation of the patient.98-100 When the patient is recently or chronically transfused, as it is typically the case in young children with congenital severe hemolytic anemia, options for phenotypic evaluation are limited. In such cases, genetic evaluation with clinically available next-generation sequencing panels may provide an accurate diagnosis necessary for appropriate management decisions.12,101,102 aHUS, atypical hemolytic uremic syndrome; HUS, hemolytic uremic syndrome; TTP, thrombotic thrombocytopenic purpura. (B) Osmotic gradient ektacytometry. The ektacytometry curve is determined by the RBC structural features.99 The points indicated in red are the following: Omin corresponds to the value of the hypotonic osmolality at which 50% of the cells hemolyze in an osmotic fragility assay and provides information on the initial surface to volume ratio of the RBCs. A shift to the right reflects a decrease in the surface area to volume ratio (ie, increased osmotic fragility). EImax corresponds to the maximum deformability of the RBC, and its value is affected by the cytoskeleton mechanics. The hypertonic descending part of the curve is represented by Ohyp, the osmolality value at which the cells’ average maximum diameter is half of EImax. The value of Ohyp correlates with the initial intracellular viscosity of the cell sample. A shift to the left reflects increased intracellular viscosity of the erythrocyte caused by increased intracellular concentration of hemoglobin, typically due to dehydration of the cell; a shift to the right may represent an overhydration state of the cell in overhydrated stomatocytosis or, most commonly, a decreased intracellular concentration of hemoglobin, such as in iron deficiency. (C) Typical osmotic gradient ektacytometry curves for various RBC membrane disorders (in red) in comparison with a normal control curve run concurrently (in green). (i) HS characterized by increased Omin and decreased EImax. (ii) HE/HPP characterized by decreased EImax and a trapezoid shape of the curve. (iii) HX with decreased Omin and decreased Ohyp. (iv) SAO with severely decreased deformability and decreased Omin.
Evaluation workflow of patients with hemolytic anemia. (A) Proposed algorithm for the laboratory evaluation of a patient presenting with symptoms and signs of hemolysis. Of note, anemia may be well compensated, as in many cases of mild HS and most cases of PIEZO1-associated HX. Evaluation for autoimmune or, especially in an infant, alloimmune hemolytic anemia with direct and indirect antiglobulin test (DAT and IAT) is the first priority, since such a diagnosis typically requires immediate action. Consideration of the possibility of microangiopathic hemolytic anemia (MAHA) and paroxysmal nocturnal hemoglobinuria (PNH) may also be necessary. Blood smear review of the patient and parents, with attention to the RBC indices, including MCV, MCHC, and red blood cell distribution width (RDW), along with hemolytic markers (bilirubin, lactate dehydrogenase, and haptoglobin [the last one reliable after 6 months of life, since earlier, it may be low due to decreased production by the infant’s liver]), ferritin, and transferrin saturation to consider iron-loading inefficient erythropoiesis, can guide the differential diagnosis. Flow cytometry with eosin-5′-maleimide (EMA) binding of band 3 and Rh-related proteins is a rapid screening test for RBC membrane disorders that are characterized by membrane loss.96,97 Osmotic fragility is increased in HS and often decreased in HX. Osmotic gradient ektacytometry, which evaluates the deformability of RBCs as they are subjected to constant shear stress in a medium of increasing osmolality in a laser diffraction viscometer, is the reference technique for differential diagnosis of erythrocyte membrane and hydration disorders when a recent transfusion does not interfere with phenotypic evaluation of the patient.98-100 When the patient is recently or chronically transfused, as it is typically the case in young children with congenital severe hemolytic anemia, options for phenotypic evaluation are limited. In such cases, genetic evaluation with clinically available next-generation sequencing panels may provide an accurate diagnosis necessary for appropriate management decisions.12,101,102 aHUS, atypical hemolytic uremic syndrome; HUS, hemolytic uremic syndrome; TTP, thrombotic thrombocytopenic purpura. (B) Osmotic gradient ektacytometry. The ektacytometry curve is determined by the RBC structural features.99 The points indicated in red are the following: Omin corresponds to the value of the hypotonic osmolality at which 50% of the cells hemolyze in an osmotic fragility assay and provides information on the initial surface to volume ratio of the RBCs. A shift to the right reflects a decrease in the surface area to volume ratio (ie, increased osmotic fragility). EImax corresponds to the maximum deformability of the RBC, and its value is affected by the cytoskeleton mechanics. The hypertonic descending part of the curve is represented by Ohyp, the osmolality value at which the cells’ average maximum diameter is half of EImax. The value of Ohyp correlates with the initial intracellular viscosity of the cell sample. A shift to the left reflects increased intracellular viscosity of the erythrocyte caused by increased intracellular concentration of hemoglobin, typically due to dehydration of the cell; a shift to the right may represent an overhydration state of the cell in overhydrated stomatocytosis or, most commonly, a decreased intracellular concentration of hemoglobin, such as in iron deficiency. (C) Typical osmotic gradient ektacytometry curves for various RBC membrane disorders (in red) in comparison with a normal control curve run concurrently (in green). (i) HS characterized by increased Omin and decreased EImax. (ii) HE/HPP characterized by decreased EImax and a trapezoid shape of the curve. (iii) HX with decreased Omin and decreased Ohyp. (iv) SAO with severely decreased deformability and decreased Omin.
Extensive studies on RBC membrane physiology and pathology in humans and mouse models for many decades have provided a comprehensive understanding of the most common erythrocyte membrane disorders, such as hereditary spherocytosis (HS), hereditary elliptocytosis (HE), and hereditary pyropoikilocytosis (HPP). The broader availability of genetic testing for clinical diagnosis (searchable in the National Center for Biotechnology Information Genetic Testing Registry: https://www.ncbi.nlm.nih.gov/gtr/) and the discovery of the molecular causes for hereditary xerocytosis (HX) have been major advances in the field over the last decade, improving feasibility of accurate diagnosis and revealing that the incidence of less common RBC membrane disorders may be underestimated. We will review here the genotypic and phenotypic variability of the RBC membrane disorders with the goal to raise the index of suspicion and emphasize the need for correct, timely diagnosis.
RBC cytoskeleton disorders
HS
Pathophysiology and genetics
HS is the most common RBC membrane disorder worldwide and the most common hereditary hemolytic anemia (HHA) in people of Northern European ancestry, with prevalence of 1 in 1000 to 2500.12-14 Spherocytes are formed due to loss of RBC membrane because of inadequate vertical linkages between the cytoskeleton and the lipid bilayer.13 Specifically, the molecular mechanism causing HS is quantitative deficiency in α- or β-spectrin, ankyrin, protein 4.2, or band 3, coded by the genes SPTA1, SPTB, ANK1, EPB42, and SLC4A1, respectively.3 An abundance of family-private mutations have been found in each of these genes to cause HS. Loss of membrane leads to decreased cell surface area to volume ratio, mandating a spherical shape that is associated with increased osmotic fragility and decreased deformability. The result is extravascular hemolysis due to increased destruction of RBCs as they pass through the spleen.
Heterozygous mutations of ANK1, SLC4A1, and SPTB genes cause autosomal dominant (AD) HS, which accounts for approximately two-thirds of the cases.13,15 10% to 15% of HS cases are inherited in an autosomal recessive (AR) fashion, caused by biallelic defects in EPB42, SPTA1, or ANK1.12,13,16 De novo mutations have also been described, most commonly for the ANK1 gene, likely due to high G+C content predisposing to slipped strand mispairing during DNA replication.17
AR EPB42-associated HS is due to complete or near-complete absence of protein 4.2 leading to band 3 decrease in the RBC membrane; heterozygous carriers of EPB42-HS are asymptomatic. This type of HS is usually of mild or moderate severity and accounts for 40% to 50% of HS in Japan but <5% of all cases in other populations.18,19 Patients with AR HS due to α-spectrin deficiency typically have a null (eg, nonsense or frameshift) SPTA1 mutation in trans to a low-expression allele with an intronic variant that activates alternative splicing, most commonly the variant c.4339-99C>T called αLEPRA (low-expression PRAgue).20-22 αLEPRA has a minor allele frequency (MAF) of 0.49% (gnomad.broadinstitute.org) and produces only ∼16% of full-length spectrin as compared with the normal SPTA1 allele or ∼8% in comparison with 2 normal SPTA1 alleles.20 This rather low amount of α-spectrin allows for a normalized RBC survival in patients with AR SPTA1-associated HS after splenectomy, pointing also to the fact that 2 normal SPTA1 alleles produce redundant levels of protein (specifically, α-spectrin is produced in threefold excess of β-spectrin).23 Complete deficiency of α-spectrin, due to biallelic null SPTA1 mutations, leads to the most severe form of HS presenting as fatal hydrops fetalis in a term pregnancy.22 Babies with such genotype may survive if they are delivered prematurely, allowing for early transfusion support, or receive intrauterine transfusions. Reticulocytopenia is characteristic of this rare type of HS; splenectomy reveals reticulocytosis but fails to significantly improve the extremely decreased survival of the spectrin-deficient RBCs.22 The parents of patients with SPTA1-associated HS, even when carrying 1 null SPTA1 allele, are asymptomatic22,24 ; some may have slight reticulocytosis or increased osmotic fragility in incubated blood.13 AR ANK1-associated HS is usually due to a null mutation in trans to a promoter or intronic variant affecting levels of expression or a missense mutation decreasing, but not obliterating, the incorporation of ankyrin into the cytoskeleton; these patients have typically severe, transfusion-dependent disease responding to splenectomy.15 A handful of cases with homozygous SLC4A125,26 variants presenting with life-threatening hydrops fetalis and remaining transfusion dependent even after splenectomy have also been reported. Homozygous, truly null SPTB mutations have not been described, indicating that complete deficiency of β-spectrin may be incompatible with life.
Clinical picture and management
The phenotypic variability of HS matches the variability in the associated causative genetic variants. Interestingly, patients with the same genotype may also have differences in their phenotype. Heterogeneity in the level of expression from splicing variants, variability in erythropoiesis response, and sometimes a different tolerance of anemia by patients and/or parents may contribute to this phenomenon of apparent incomplete penetrance. Patients with AD SLC4A1, SPTB, ANK1, and AR EPB42-associated HS present with a range of well-compensated hemolysis up to a moderately severe anemia (hemoglobin <8 g/dL with brisk reticulocytosis >10%).13,19 AR ANK1- or SPTA1-associated HS is most frequently a transfusion-dependent disease since early infancy.22 All types of HS are likely to present with neonatal jaundice within the first 24 hours of life, posing risk of kernicterus without appropriate monitoring and treatment with phototherapy; rarely, exchange transfusion is also needed.27 After 2 to 3 weeks, when the normal neonatal hyposplenism resolves, anemia ensues and may be severe enough to require transfusion. Transfusion dependency during the first 9 months of life is not rare with any type of HS, due to the delayed erythropoietin response, and is not prognostic of the severity of the disease.28 Infants with frequent transfusion requirement despite evidence by family history or genetic evaluation that they have an AD form of HS may benefit from erythropoietin administration.29
Patients with compensated hemolysis may present at a time of hemolytic or aplastic crisis due to a concurrent viral infection (eg, Epstein-Barr virus or parvovirus B19, respectively) or with gallstones at a relatively young age. Jaundice and gallstones may be exacerbated with coinheritance of a bilirubin metabolism defect, such as Gilbert syndrome.30 Examples of red cell morphology per gene affected13,31 and associated ektacytometry are depicted in Figure 3. Mean cellular volume (MCV) is usually normal, while mean corpuscular hemoglobin concentration (MCHC) is increased at high-normal or above-normal (>36g/dL) values. Since decreased deformability of spherocytes leads to their increased destruction in the spleen, splenectomy improves anemia. Notable exceptions are the rare cases due to near-complete deficiency of α-spectrin or band 3, which with the advent of intrauterine transfusions can survive.22,26 These patients require either a lifelong program of transfusions and iron chelation or stem cell transplant. Splenectomy is indicated for HS patients with moderate and severe anemia requiring frequent transfusions and/or having compromised quality of life. Due to the lifelong risk of sepsis after splenectomy, the patient should be immunized against encapsulated bacteria and receive immediate medical attention with blood culture and IV antibiotics for fever. Partial splenectomy (removal of 80%-90% of the spleen) is also effective to mitigate hemolysis and offers the advantage of maintaining splenic immune function, which is especially important for cases in which splenectomy is performed at age <5 years.12,32-34
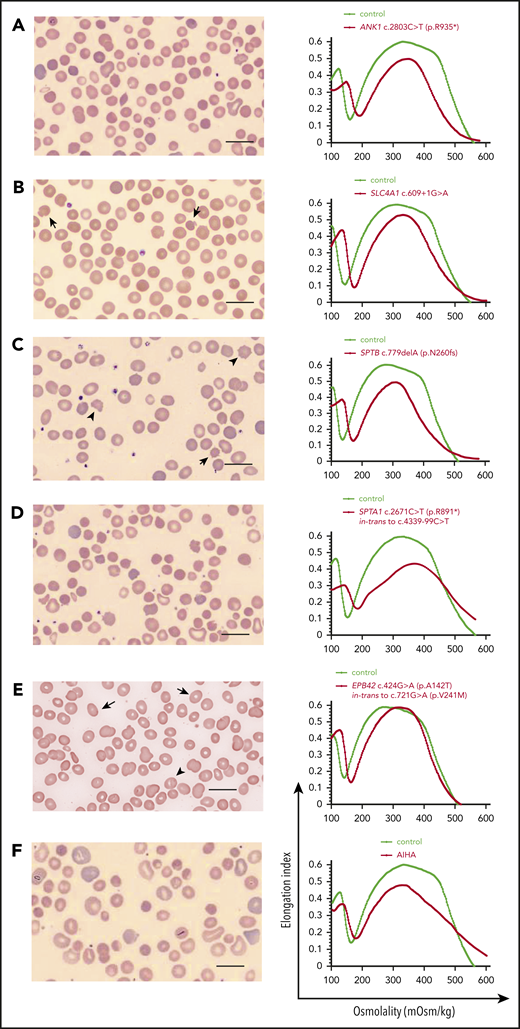
HS. Examples of red cell morphology in HS due to different gene mutations and associated osmotic gradient ektacytometry curves. Spherocytes (ie, RBCs with no or decreased central pallor) predominate, but additional RBC morphology characteristics may provide hints to the gene/protein defect causing HS, as Palek and Sahr31 and Eber and Lux13 have described very astutely in the past. (A-E, left) Blood smear of a patient with AD ANK1-HS showing anisocytosis (A). Occasional mushroom-shaped red cells (arrows) are characteristic of HS due to deficiency of band 3 (SLC4A1) (B). Acanthocytes (arrowheads) and echinocytes (arrow) are noted along with spherocytes in SPTB-associated HS (C). Blood smear of a not-recently transfused patient with AR SPTA1-HS demonstrates remarkable anisocytosis and poikilocytosis with contracted dense cells (D). Ovalocytes (arrows) and few ovalostomatocytes (arrowheads) are noted in AR EPB42-HS (E). Nevertheless, the above described changes in RBC morphology are not always specific for the gene affected in HS.102 Scale bars, 14 µm; Wright-Giemsa stain. (A-E, right) The ektacytometry curve in HS is characterized by increased Omin, which corresponds to the increased osmotic fragility of the spherocytes. In almost all cases, decreased maximum deformability indicated by low EImax is also noted, as well as decreased Ohyp. The decrease in EImax tends to correlate with the degree of hemolysis and severity of anemia. (F) Important for differential diagnosis is that not all spherocytosis is hereditary. Autoimmune hemolytic anemia due to warm-reacting immunoglobulin G causes RBC membrane loss and acquired spherocytosis, with erythrocyte morphology and ektacytometry resembling HS. It is advisable to initiate the workup by first considering the possibility of an immune-mediated cause in every hemolytic anemia, since this radically alters management. In addition, blood smear and ektacytometry in congenital dyserythropoietic anemia type II also resemble HS; higher MCV, suboptimal reticulocytosis, and ferritin values disproportionally high for the number of transfusions will provide a clue to this possibility if bone marrow studies have not yet been performed. AIHA, autoimmune hemolytic anemia.
HS. Examples of red cell morphology in HS due to different gene mutations and associated osmotic gradient ektacytometry curves. Spherocytes (ie, RBCs with no or decreased central pallor) predominate, but additional RBC morphology characteristics may provide hints to the gene/protein defect causing HS, as Palek and Sahr31 and Eber and Lux13 have described very astutely in the past. (A-E, left) Blood smear of a patient with AD ANK1-HS showing anisocytosis (A). Occasional mushroom-shaped red cells (arrows) are characteristic of HS due to deficiency of band 3 (SLC4A1) (B). Acanthocytes (arrowheads) and echinocytes (arrow) are noted along with spherocytes in SPTB-associated HS (C). Blood smear of a not-recently transfused patient with AR SPTA1-HS demonstrates remarkable anisocytosis and poikilocytosis with contracted dense cells (D). Ovalocytes (arrows) and few ovalostomatocytes (arrowheads) are noted in AR EPB42-HS (E). Nevertheless, the above described changes in RBC morphology are not always specific for the gene affected in HS.102 Scale bars, 14 µm; Wright-Giemsa stain. (A-E, right) The ektacytometry curve in HS is characterized by increased Omin, which corresponds to the increased osmotic fragility of the spherocytes. In almost all cases, decreased maximum deformability indicated by low EImax is also noted, as well as decreased Ohyp. The decrease in EImax tends to correlate with the degree of hemolysis and severity of anemia. (F) Important for differential diagnosis is that not all spherocytosis is hereditary. Autoimmune hemolytic anemia due to warm-reacting immunoglobulin G causes RBC membrane loss and acquired spherocytosis, with erythrocyte morphology and ektacytometry resembling HS. It is advisable to initiate the workup by first considering the possibility of an immune-mediated cause in every hemolytic anemia, since this radically alters management. In addition, blood smear and ektacytometry in congenital dyserythropoietic anemia type II also resemble HS; higher MCV, suboptimal reticulocytosis, and ferritin values disproportionally high for the number of transfusions will provide a clue to this possibility if bone marrow studies have not yet been performed. AIHA, autoimmune hemolytic anemia.
HE and HPP
Pathophysiology and genetics
HE-causing mutations are common in malaria-endemic regions, with a prevalence of 0.6% to 3% in West Africa, supporting the hypothesis that elliptocytosis confers survival advantage to malaria. In the United States, the incidence of HE is 1 in 2000 to 4000 people, although this may be underestimated, since HE is frequently asymptomatic, with no or mild hemolytic anemia. It is caused by monoallelic (heterozygous) mutations in the genes encoding α-spectrin (SPTA1), β- spectrin (SPTB), and protein 4.1R (EPB41). HE-causing SPTA1 and SPTB variants are located in the spectrin tetramerization domain (Figure 1B) or distally and produce altered spectrin chains that incorporate into the cytoskeleton and weaken the spectrin tetramer.35 EPB41 mutations lead to altered or deficient protein 4.1R that compromises the spectrin-4.1R-actin association at the junctions.12,36 Rare biallelic mutations of GYPC causing complete deficiency of glycophorin C (Leach phenotype) also cause HE due to concurrent partial deficiency of 4.1R, with no significant hemolysis.37 The defective “horizontal” linkages in the cytoskeleton, caused by HE mutations, affect RBC membrane deformability and shape, allowing permanent deformation under shear stress into elliptocytes.36 The most common form of HPP, frequently diagnosed in infants of African ancestry presenting with neonatal hyperbilirubinemia (Figure 4), is due to an SPTA1 HE-causing mutation in trans to the intronic SPTA1 variant c.6531-12C>T known as αLELY (low-expression LYon).38,39 αLELY is a common variant (MAF 25.5%) (gnomad.broadinstitute.org), leading to a 50% decrease in α-spectrin expression. αLELY is clinically silent in normal individuals, even in the homozygous state, since α-spectrin is produced in excess, but in trans to an HE-causing SPTA1 mutation leads to aggravation of the phenotype to HPP, because it allows for increased relative incorporation of the abnormal spectrin chain into the cytoskeleton. HPP can also be caused by biallelic HE-causing mutations, homozygous or compound heterozygous variants of SPTA1, SPTB, EPB41, or an allele each of SPTA1 and SPTB carrying HE mutations.36,38,40
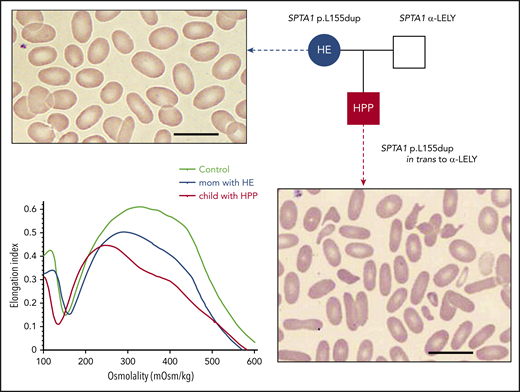
HE and HPP. The most common form of HPP, diagnosed in infants with neonatal hyperbilirubinemia, is due to an SPTA1 HE-causing mutation in trans to the intronic SPTA1 variant c.6531-12C>T known as αLELY (low-expression LYon). Familial studies at the time reveal that 1 parent carries the SPTA1 HE-causing mutation and has elliptocytosis (smear characterized by elliptically shaped red blood cells), while the other parent carries αLELY with a normal erythrocyte phenotype. The blood smear in HPP is characterized by marked anisocytosis and poikilocytosis with bizarre microcytes and fragmented cells along with elliptocytes (scale bars, 14 µm; Wright-Giemsa stain). HPP RBCs were found early on to have increased thermal sensitivity; this is maybe the source of the name pyro (coming from the Greek word “πυρ” meaning fire)-poikilocytosis. Another possibility for the origin of the term is that the cells resemble the morphology of the blood smear in patients with the microangiopathic hemolytic anemia of patients with extensive burns.
HE and HPP. The most common form of HPP, diagnosed in infants with neonatal hyperbilirubinemia, is due to an SPTA1 HE-causing mutation in trans to the intronic SPTA1 variant c.6531-12C>T known as αLELY (low-expression LYon). Familial studies at the time reveal that 1 parent carries the SPTA1 HE-causing mutation and has elliptocytosis (smear characterized by elliptically shaped red blood cells), while the other parent carries αLELY with a normal erythrocyte phenotype. The blood smear in HPP is characterized by marked anisocytosis and poikilocytosis with bizarre microcytes and fragmented cells along with elliptocytes (scale bars, 14 µm; Wright-Giemsa stain). HPP RBCs were found early on to have increased thermal sensitivity; this is maybe the source of the name pyro (coming from the Greek word “πυρ” meaning fire)-poikilocytosis. Another possibility for the origin of the term is that the cells resemble the morphology of the blood smear in patients with the microangiopathic hemolytic anemia of patients with extensive burns.
Clinical picture and management
HE is associated with low rate of hemolysis, with normal hemoglobin values and borderline high reticulocyte count. It is frequently diagnosed during investigation for gallstones or splenomegaly or in family work-up in a parent of an infant with neonatal jaundice diagnosed with HPP. Infants with the common form of HPP (SPTA1 HE-variant in trans to αLELY) may have moderate to severe hemolytic anemia requiring frequent transfusions early in life, but the phenotype improves after the first 1 to 2 years and evolves into typical HE.12 HPP due to biallelic HE-causing mutations may present with mild to severe chronic anemia. Most of the cases with severe hemolysis and chronic transfusion requirement respond to splenectomy,38,40 but rare cases have persistent severe hemolysis even after splenectomy, especially if there is a concurrent HS component of decreased protein expression. For example, splenectomy failed to improve transfusion dependency in a child with compound heterozygosity for a previously reported splice site SPTA1 mutation (c.2806-13T>G) known as spectrin St. Claude41 and a novel nonsense SPTA1 mutation (c.352C>T; p.R118*) in trans, predicted to be associated with severe HPP due to incorporation in the membrane of only the truncated α-spectrin produced by the spectrin St. Claude allele (T.A.K., unpublished observation).
SAO
Southeast Asian ovalocytosis (SAO) is an AD HHA caused by deletion in SLC4A1 of 27 base pairs encoding amino acids 400 to 408 of band 3 at the junction of the N-terminal cytoplasmic domain with the transmembrane domain, resulting in abnormal folding of the protein.42,43 SAO confers resistance to cerebral malaria44 and has been under selective advantage in the Pacific rim of Southeast Asia, with MAF in this population ≤1%. Newborns with SAO frequently present with neonatal hyperbilirubinemia.45 Hemolysis resolves completely by 3 years of age, and SAO heterozygotes are asymptomatic, despite their large, oval-shaped RBCs, which are markedly rigid.46 SAO homozygosity is lethal in utero with no intervention. Only 1 homozygous patient has been described presenting with hydrops fetalis at 22 weeks of gestation, salvaged with intrauterine transfusions, and born with transfusion-dependent, hemolytic, and dyserythropoietic anemia and distal renal tubular acidosis.47
RBC hydration disorders
Dehydrated hereditary stomatocytosis or HX
Pathophysiology and genetics
HX is a clinically heterogeneous, AD HHA caused by mutations in PIEZO1,48-50 a mechanosensitive nonselective cation channel (Figure 5A-B), or, less commonly, by mutations in the Ca2+-activated K+ channel KCNN4 (Gardos channel).51-55 It is characterized by an abnormal RBC K+ leak not compensated by a proportional intracellular Na+ gain, leading to cellular dehydration. Although HX due to PIEZO1 mutations and HX due to mutation in KCNN4 share many characteristics, including a variable degree of RBC dehydration, hemolysis that persists after splenectomy, and iron overload disproportionate to the transfusion history,56 there are differences in cellular pathophysiology, clinical presentation, and potential response to therapeutic measures, leading to suggestions to refer to KCNN4-associated HX with its own name as “Gardos channelopathy”.54,56 As RBCs navigate narrow capillaries and sinusoids, normal activation of PIEZO1 by pressure and shear stress increases intracellular Ca2+, which activates KCNN4 and causes K+ efflux and water loss, to attain a temporary decrease in cell volume and facilitate passage (Figure 5C).57-59 Most of the gain-of-function PIEZO1 mutations causing HX are due to a slower rate of channel inactivation,60,61 but other mechanisms of PIEZO1 dysfunction, including altered channel kinetics, response to osmotic stress, and membrane trafficking, have also been described and contribute to the phenotypic heterogeneity of the disease.56,62,63
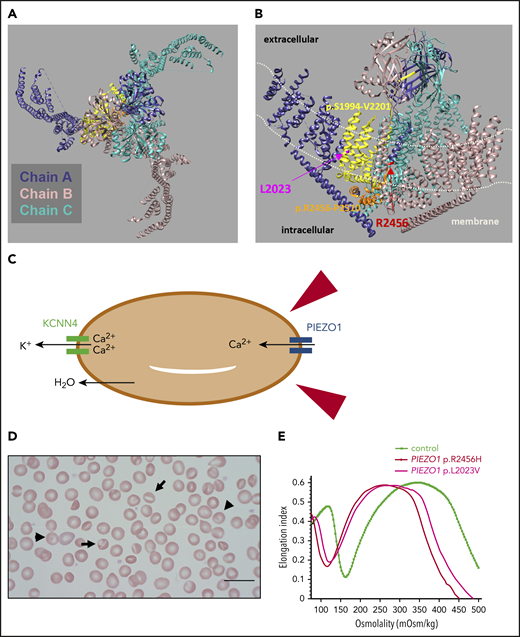
HX due to heterozygous PIEZO1 mutation. Human PIEZO1 is a 2521 amino acid (287 kDa) protein with ∼38 transmembrane helices. Cryoelectron microscopy studies of the highly homologous mouse Piezo1 reveal that PIEZO1 trimers form an elegant 3-bladed propeller structure with a curved transmembrane region creating an inverted membrane dome and a central ion pore formed by the C-terminal domains of the subunits, in which most of the disease-causing mutations are located.103-106 This unique structure of PIEZO1 senses changes in membrane tension to alter gating of the ion channel.106,107 (A) Top view of the homotrimeric PIEZO1 channel showing its 3-bladed, propeller-like architecture (from the extracellular space looking down through the central pore). The 3 subunits of the trimer are color coded. (B) Side view of the homotrimeric PIEZO1 channel showing the curved transmembrane region, which creates a membrane dome.106,107 The position of the membrane is roughly indicated with white dotted lines. Two of the mutation “hot spot” areas described by Picard et al56 are highlighted in chain A (dark blue); the sequence p.R2456-P2510 in the pore domain (coded within exon 51) is colored orange, and one of the most common HX mutations in this region (p.R2456) is labeled in red; the sequence p.S1994-V2201 (coded within exons 42-45) is colored yellow, and one of the mutations in this area (p.L2023V) is indicated in magenta. A third hot-spot region for mutations is located N-terminally in exons 14 to 18; the structure of this area has not yet been modeled. Patients demonstrating a more severe phenotype are more likely to have mutations in the PIEZO1 pore domain.56 Images created using UCSF Chimera,108 with the Protein Data Bank structure model 3JAC. (C) Sketch of interaction between PIEZO1 and KCNN4 as RBCs travel through the vasculature. In narrow capillaries and sinusoids, mechanical stress (represented by the red arrowheads) results in activation of PIEZO1 and Ca2+ entry. Increased intracellular Ca2+ leads to activation of KCNN4 (a calcium ion binds to each of the 4 calmodulin molecules tightly bound to the cytoplasmic domains of the 4 KCNN4 subunits),109 and K+ efflux ensues. Subsequent water loss results in a temporary decrease in cell volume and facilitates passage.57-59 (D) Blood smear from a patient heterozygous for p.R2456H with macrocytosis (MCV 96 fL) showing occasional stomatocytes (arrows), target cells (arrowheads), and dense cells (thin arrows). (E) Osmotic gradient ektacytometry showing the typical HX curve with left shift due to decreased Omin and Ohyp, indicating RBC dehydration. Ekatacytometry profiles are shown for 2 patients with HX due to PIEZO1 p.R2456H and p.L2023V.
HX due to heterozygous PIEZO1 mutation. Human PIEZO1 is a 2521 amino acid (287 kDa) protein with ∼38 transmembrane helices. Cryoelectron microscopy studies of the highly homologous mouse Piezo1 reveal that PIEZO1 trimers form an elegant 3-bladed propeller structure with a curved transmembrane region creating an inverted membrane dome and a central ion pore formed by the C-terminal domains of the subunits, in which most of the disease-causing mutations are located.103-106 This unique structure of PIEZO1 senses changes in membrane tension to alter gating of the ion channel.106,107 (A) Top view of the homotrimeric PIEZO1 channel showing its 3-bladed, propeller-like architecture (from the extracellular space looking down through the central pore). The 3 subunits of the trimer are color coded. (B) Side view of the homotrimeric PIEZO1 channel showing the curved transmembrane region, which creates a membrane dome.106,107 The position of the membrane is roughly indicated with white dotted lines. Two of the mutation “hot spot” areas described by Picard et al56 are highlighted in chain A (dark blue); the sequence p.R2456-P2510 in the pore domain (coded within exon 51) is colored orange, and one of the most common HX mutations in this region (p.R2456) is labeled in red; the sequence p.S1994-V2201 (coded within exons 42-45) is colored yellow, and one of the mutations in this area (p.L2023V) is indicated in magenta. A third hot-spot region for mutations is located N-terminally in exons 14 to 18; the structure of this area has not yet been modeled. Patients demonstrating a more severe phenotype are more likely to have mutations in the PIEZO1 pore domain.56 Images created using UCSF Chimera,108 with the Protein Data Bank structure model 3JAC. (C) Sketch of interaction between PIEZO1 and KCNN4 as RBCs travel through the vasculature. In narrow capillaries and sinusoids, mechanical stress (represented by the red arrowheads) results in activation of PIEZO1 and Ca2+ entry. Increased intracellular Ca2+ leads to activation of KCNN4 (a calcium ion binds to each of the 4 calmodulin molecules tightly bound to the cytoplasmic domains of the 4 KCNN4 subunits),109 and K+ efflux ensues. Subsequent water loss results in a temporary decrease in cell volume and facilitates passage.57-59 (D) Blood smear from a patient heterozygous for p.R2456H with macrocytosis (MCV 96 fL) showing occasional stomatocytes (arrows), target cells (arrowheads), and dense cells (thin arrows). (E) Osmotic gradient ektacytometry showing the typical HX curve with left shift due to decreased Omin and Ohyp, indicating RBC dehydration. Ekatacytometry profiles are shown for 2 patients with HX due to PIEZO1 p.R2456H and p.L2023V.
Human KCNN4 has 6 transmembrane helices and forms a tetramer in the RBC membrane.64 KCNN4 mutations affect channel kinetics displaying increased current magnitude, like PIEZO1 gain-of-function mutations. However, the cellular pathology may be more complex with compensatory effects by multiple ion-transport systems in patients with HX due to KCNN4 p.R352H54 and paradoxically reduced activity of the p.V282M mutant channel under conditions of maximal stimulation.55 A 28-base-pair deletion, predicted to affect the KCNN4-calmodulin binding domain, has also been described.56
HX is often described as a rare condition, but there are several lines of evidence that indicate that it is rather underdiagnosed.65 Estimates based on reported cases suggest ∼1 case of HX in 50 000 births.66 However, a recent study performed using a large US commercial laboratory database to search for complete blood count results consistent with HX estimated an incidence of ∼1 case in 8000 adults.67 In addition, a recent study described a PIEZO1 gain-of-function polymorphism (E756del) predicted to be present in ≥1 allele in approximately one-third of the African population and demonstrated that individuals heterozygous for this variant have more dense RBCs that are less susceptible to infection by the malaria parasite Plasmodium falciparum.68 It was recently shown that this variant may also be a disease-modifying polymorphism for individuals with sickle cell disease.69
Clinical picture and management
The high degree of variability in the HX phenotype probably has many causes, including a large number of causative variants, with variable mechanisms of dysfunction; differences in transporters or other proteins that function in concert with the mutated protein; and coinheritance of other gene variants affecting RBC phenotype. HX patients typically present with mild to moderate hemolytic anemia or fully compensated hemolysis; a few cases with polycythemia (hemoglobin >16 g/dL) despite hemolysis have been reported.56,70 Many patients may not come to medical attention until adulthood. Patients with HX due to PIEZO1 variants typically have high reticulocyte count, high or high-normal MCHC, high MCH, and high MCV.71 Peripheral blood smears reveal occasional stomatocytes, target cells, and possibly dense cells (Figure 5B). Splenomegaly is common. Some patients have history of transient perinatal edema or nonimmune hydrops fetalis. Total bilirubin is frequently increased, and many patients develop gallstones. Ferritin levels are typically elevated, and iron overload is disproportionate to transfusion history. Some individuals present with a primary clinical sign of hepatic and/or myocardial iron overload.63 Osmotic fragility is frequently, but not always, decreased, limiting its value as a diagnostic test in these cases. Osmotic gradient ektacytometry typically shows a leftward shift of the curve, indicating dehydration (Figure 5D). RBC intracellular cation determinations reveal reduced K+ content without corresponding increase in Na+ content.70
In the largest study of HX to date, individuals with KCNN4-HX had more significant anemia, with lower reticulocyte count, and higher ferritin values consistent with worse iron overload in comparison with PIEZO1-HX.56 Ektacytometry curve was dramatically left-shifted with reduced EImax and Ohyp values consistent with severe cell dehydration in HX due to KCNN4 p.V282M55 but was near normal, with no clear evidence of cellular dehydration in HX due to KCNN4 p.R352H, likely because of compensatory effects including K+ influx through KCC, NKCC and Na/K ATPase channels.54,56,72
Gene sequencing with appropriately designed genetic panels is essential for an accurate diagnosis. Several patients with HX have received the diagnosis of HS or congenital dyserythropoietic anemia prior to genetic evaluation. Dysplastic and binucleated erythroblasts can be seen with stress erythropoiesis associated with brisk hemolytic anemia, while there may also be direct effects of abnormal PIEZO1 activation in erythropoiesis, as it was recently shown to delay erythroid maturation73 and impair reticulocyte maturation74 in vitro.
Management is supportive for anemia and complications of hemolysis with vigilant monitoring for iron overload. Ferritin and transferrin saturation should be monitored, and T2* magnetic resonance imaging of the liver and heart is indicated when iron overload is suspected. Poor correlation between ferritin level and mean liver iron content was shown for individuals with a ferritin level <1000 ng/mL, suggesting the value of evaluating iron content by imaging in patients with even moderate ferritin increases at diagnosis.56 Phlebotomy or iron chelation are being used to address HX-associated iron overload, which can lead to cirrhosis and/or heart failure if left untreated.56 A comparative efficacy study would be valuable, since the driver for hemochromatosis in HX may be the excessive erythropoiesis causing increased erythroferrone that suppresses hepcidin.75 Therefore, the efficacy of phlebotomy to treat iron overload in HX, while it may further stimulate erythropoiesis, needs evaluation.76 Splenectomy is strictly contraindicated in PIEZO1-associated HX. It is not of therapeutic benefit in reducing hemolysis, which is largely intravascular in HX, and results in a greatly increased risk of life-threatening venous and arterial thromboembolic complications,77,78 which have been shown to happen within 1 month up to 28 years after splenectomy.56 Interestingly, thrombotic complications were not observed in a total of 12 KCNN4-HX splenectomized patients after a follow-up time of 2 to 44 years.54,56 These patients had well-compensated cellular dehydration based on MCHC and/or ektacytometry studies and also maintained active volume regulation with an overactivated Gardos channel; nevertheless, they continued to have significant hemolysis, even after splenectomy.54,56 Because perinatal edema and nonimmune hydrops fetalis are common in HX, pregnancies should be closely monitored when either parent has HX.56
Specific drugs for the treatment of HX are not currently available. However, because KCNN4 has been demonstrated to be the major mediator of RBC dehydration in HX due to mutated PIEZO1,79 a specific KCNN4 inhibitor, senicapoc, has been suggested as a candidate for treatment of HX due to either PIEZO1 or KCNN4 mutation. In a phase 3 clinical trial for the use of senicapoc for sickle cell disease, it reduced RBC dehydration and hemolysis, increased hemoglobin levels, and was well tolerated.80 It has also been shown to inhibit both wild-type and mutated KCNN4 channels and reduce RBC dehydration in vitro.81 Although small-molecule agonists have been useful in the study of PIEZO1 function, specific small-molecule inhibitors for PIEZO1 have not yet been identified, and it will likely be challenging to achieve the desired therapeutic improvement in RBC hydration by inhibiting PIEZO1 without affecting the wide range of its functions in various other cell types.82
Overhydration syndromes
Pathophysiology and genetics
RBC overhydration syndromes are characterized by a large net increase in RBC intracellular Na+ and water not adequately compensated by a decrease in intracellular K+, leading to an increase in RBC volume without corresponding increase in membrane surface area.83,84
Overhydrated hereditary stomatocytosis (OHSt), caused by heterozygous missense mutations in RHAG, is a rare AD HHA. RHAG codes for Rhesus-associated glycoprotein (RhAG), which functions as an ammonium transporter,85 and is a component of the band 3 macromolecular complex (Figure 1B). OHSt-associated mutant RhAG proteins expressed in Xenopus oocytes induce monovalent cation leak several-fold greater than the wild-type RhAG, probably by altering a cytoplasmic constriction in the pore of the protein.86
Cryohydrocytosis (CHC) is caused by certain heterozygous missense mutations in SLC4A1 causing minimally increased RBC cation leak at physiologic temperature but a major leak as temperature approaches 5°C.84 The CHC mutations are located at the band 3 transmembrane segment involved in anion exchange, altering it to mediate cation leak.87 This leak can be inhibited by specific band 3 inhibitors in vitro.88 In some cases, part of the observed cation leak may be mediated through band 3 regulation of other transporters.89 Individuals with SLC4A1 mutations generally have a milder form of OHSt than individuals with RhAG mutations.65
Another rare form of CHC, referred to as stomatin-deficient CHC, is caused by mutations in SLC2A1 coding for GLUT1, the glucose transporter that also binds stomatin.90-92 In stomatin-deficient CHC, there is a deficiency in GLUT1 and stomatin and a large RBC cation leak that increases in the cold. In addition to moderate hemolytic anemia, individuals with these mutations have cataracts due to the altered cation permeability and neurological disorder (seizures, mental retardation, dyskinesias) related to inadequate glucose transport into neurons.
Other band 3 disorders that result in some degree of RBC overhydration include SAO and band 3 Ceinge. SAO RBCs were described above with the cytoskeleton disorders but have also been shown to have a cation leak in the cold similar to that observed in CHC mutations.46 Band 3 Ceinge (SLC4A1 p.Gly796Arg) is an unusual, dominantly inherited variant associated with mild hemolytic anemia with dyserythropoiesis.93 The mutation is located in the same membrane-spanning domain as many CHC mutants and has a similar cation leak at low temperature. Since membrane cation transport pathways are regulated by phosphorylation-dephosphorylation events, the RBC membrane tyrosine phosphorylation profile was evaluated, revealing membrane association of the Syk and Lyn tyrosine kinases and tyrosine phosphorylation of band 3 and stomatin.93
Clinical picture and management
Patients with overhydration syndromes may have a mild to severe hemolytic anemia. Stomatocytes are usually abundant on peripheral blood smears along with occasional spherocytes. MCV is typically increased and MCHC decreased. Osmotic fragility is increased, and the osmotic gradient ektacytometry curve is shifted to the right. Intracellular cation determinations reveal greatly increased RBC Na+ content and reduced K+ content.
Management is supportive for anemia and complications of hemolysis. Splenectomy is partially effective in reducing hemolysis, but it carries a high risk of thromboembolic complications and pulmonary hypertension.77,78
Conclusions
The broader availability of next-generation sequencing over the last decade has vastly facilitated accurate diagnosis in patients with HHA and has allowed genotype-phenotype correlations and natural history studies in patient cohorts with rare but well-defined pathogenesis. These studies have revealed complications such as iron overload in HX, amenable to monitoring, treatment, and prevention, and made clear that the one-size-fits-all approach of splenectomy for any patient with jaundice is not always effective or safe. Careful phenotypic assessment in clinical or research laboratories is necessary to assist in the interpretation of variants of unknown clinical significance revealed by genetic evaluation. It is exciting to see the emerging possibility of targeted treatments to manipulate transporter activity94 or structural organization of cytoskeletal proteins via phosphorylation95 that have the potential to offer safer therapeutic options to improve morbidity and mortality.
Acknowledgments
The authors are grateful to the teams of Cincinnati Children’s Erythrocyte Diagnostic Laboratory and Molecular Genetics Laboratory for their contribution in the diagnostic studies of cases presented in the figures.
This work was supported by the National Institutes of Health, National Heart, Lung, and Blood Institute grant R01HL116352 (T.A.K.) and by the National Institutes of Health, National Center for Advancing Translational Sciences (award 1UL1TR001425-01).
The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
Authorship
Contribution: M.R. and T.A.K. wrote the paper.
Conflict-of-interest disclosure: The authors declare no competing financial interests.
Correspondence: Theodosia A. Kalfa, Cancer and Blood Diseases Institute, Cincinnati Children’s Hospital Medical Center, 3333 Burnet Ave, MLC 7015, Cincinnati, OH 45229-3039; e-mail: theodosia.kalfa@cchmc.org.

This feature is available to Subscribers Only
Sign In or Create an Account Close Modal