

Key Points
Crystal structure of the FXIIFnII domain in complex with gC1qR reveals an asymmetric interaction and bound Zn2+ ions.
gC1qR clusters FXII and kininogen into higher-order ternary complexes.

Abstract
The contact system is composed of factor XII (FXII), prekallikrein (PK), and cofactor high-molecular-weight kininogen (HK). The globular C1q receptor (gC1qR) has been shown to interact with FXII and HK. We reveal the FXII fibronectin type II domain (FnII) binds gC1qR in a Zn2+-dependent fashion and determined the complex crystal structure. FXIIFnII binds the gC1qR trimer in an asymmetric fashion, with residues Arg36 and Arg65 forming contacts with 2 distinct negatively charged pockets. gC1qR residues Asp185 and His187 coordinate a Zn2+ adjacent to the FXII-binding site, and a comparison with the ligand-free gC1qR crystal structure reveals the anionic G1-loop becomes ordered upon FXIIFnII binding. Additional conformational changes in the region of the Zn2+-binding site reveal an allosteric basis for Zn2+ modulation of FXII binding. Mutagenesis coupled with surface plasmon resonance demonstrate the gC1qR Zn2+ site contributes to FXII binding, and plasma-based assays reveal gC1qR stimulates coagulation in a FXII-dependent manner. Analysis of the binding of HK domain 5 (HKD5) to gC1qR shows only 1 high-affinity binding site per trimer. Mutagenesis studies identify a critical G3-loop located at the center of the gC1qR trimer, suggesting steric occlusion as the mechanism for HKD5 asymmetric binding. Gel filtration experiments reveal that gC1qR clusters FXII and HK into a higher-order 500-kDa ternary complex. These results support the conclusion that extracellular gC1qR can act as a chaperone to cluster contact factors, which may be a prelude for initiating the cascades that drive bradykinin generation and the intrinsic pathway of coagulation.
Introduction
The contact activation system lies at the crossroads of plasma coagulation and innate immunity and consists of proteases factor XII (FXII), prekallikrein (PK), and cofactor high-molecular-weight kininogen (HK).1-3 Binding of contact factors to the cell surface has been shown to be mediated by the complement C1q receptor (gC1qR; also known as C1QBP, HABP1, and P32) and the urokinase receptor (uPAR).4-7 gC1qR is a multicompartmental and multifunctional protein essential for mitochondrial function,8 but it is also present at the surface of stimulated cells.9-11 gC1qR has no plasma membrane anchor but forms interactions with other cell surface proteins and receptors (cytokeratin-1, β1-integrin, dendritic cell–specific ICAM-3–grabbing nonintegrin receptor, and fibrinogen).4,12-15 The gC1qR crystal structure is a doughnut-shaped symmetrical trimer with both a highly acidic ligand-binding surface and a cell-binding face.16 gC1qR has been characterized as binding a diverse array of structurally distinct ligands7,9,15,17,18 and has been proposed to function as a chaperone directing the assembly of multiprotein complexes.19
The domain structures of FXII, HK, and gC1qR are shown in Figure 1A. Crystal structures have been determined for the FXII protease domain20-22 and the fibronectin type I (FnI) and epidermal growth factor-like domains.23 As the N-terminal FXIIFnII domain is central to understanding processes of FXII conformational regulation24 we determined the crystal structure in complex with gC1qR. This revealed an asymmetric FXII-binding mode and a novel Zn2+-binding site in the gC1qR structure.
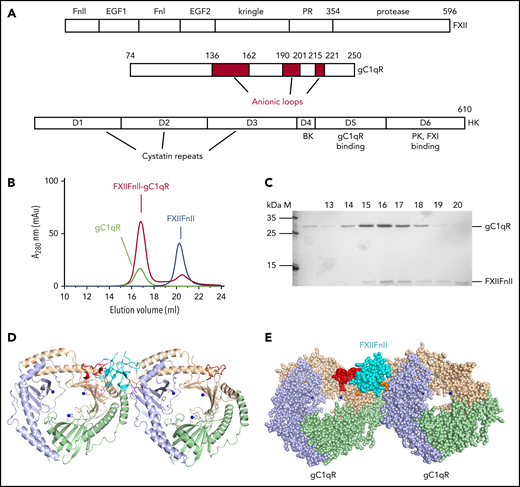
Structure of FXII, gC1qR, and HK. (A) Domain organization of FXII, gC1qR, and HK. The gC1qR anionic loops are red and residue numbers for domain boundaries are labeled. HK domain D4 represents the bradykinin (BK) peptide. (B) Gel filtration elution profiles for FXIIFnII (blue) and gC1qR (black) in isolation and a gC1qR-FXIIFnII mixture (red). (C) Coomassie-stained SDS-PAGE gel of the fractions collected from gel filtration of the FXIIFnII-gC1qR mixture. Lanes labeled 15 to 18 correspond to elution volumes 15 and 18 mL from panel B showing both proteins coeluting as a complex with excess FXIIFnII observed at 20 mL. The first lane (labeled M) is the protein marker, and the second lane is gC1qR in isolation. (D) Diagram of the FXIIFnII-gC1qR complex crystal structure showing the FXIIFnII domain (cyan) in complex with gC1qR (wheat, purple, and green), with Zn2+ in blue, the anionic G1-loop in red, and key interacting side chains as sticks. (E) Space-filling representation of the gC1qR complex with the FXIIFnII domain as spheres (cyan), with residues Arg36 and Arg65 in orange.
Structure of FXII, gC1qR, and HK. (A) Domain organization of FXII, gC1qR, and HK. The gC1qR anionic loops are red and residue numbers for domain boundaries are labeled. HK domain D4 represents the bradykinin (BK) peptide. (B) Gel filtration elution profiles for FXIIFnII (blue) and gC1qR (black) in isolation and a gC1qR-FXIIFnII mixture (red). (C) Coomassie-stained SDS-PAGE gel of the fractions collected from gel filtration of the FXIIFnII-gC1qR mixture. Lanes labeled 15 to 18 correspond to elution volumes 15 and 18 mL from panel B showing both proteins coeluting as a complex with excess FXIIFnII observed at 20 mL. The first lane (labeled M) is the protein marker, and the second lane is gC1qR in isolation. (D) Diagram of the FXIIFnII-gC1qR complex crystal structure showing the FXIIFnII domain (cyan) in complex with gC1qR (wheat, purple, and green), with Zn2+ in blue, the anionic G1-loop in red, and key interacting side chains as sticks. (E) Space-filling representation of the gC1qR complex with the FXIIFnII domain as spheres (cyan), with residues Arg36 and Arg65 in orange.
Materials and methods
Protein expression and purification
The human gC1qR (residues 74-282 with Leu74 substituted to Met74) Escherichia coli expression vector was a gift from Adrian R. Krainer (Cold Spring Harbor Laboratory). Expression and purification of gC1qR was performed as described previously.16 gC1qR variants were generated by site-directed mutagenesis using the Agilent Technologies QuikChange Kit. HKD5 was expressed in E. coli using the pET28a vector, and D5-1, D5-2, and HK 401-438 were expressed as glutathione S-transferase fusion proteins using the pGEX 4T-1 vector (supplemental Figure 1, available on the Blood Web site). Untagged FXIIFnII domain (residues 1-71) was cloned into vector pMT-PURO for expression in the insect cell–based DES system (Invitrogen) using previously described protocols.22 FXIIFnII was initially purified from media using a Capto-S column (GE Healthcare) equilibrated with 0.05 M 2-(N-morpholino)ethanesulfonic acid (pH 6.0), and a gradient of 0 to 1.0 M NaCl was used for elution. Subsequently, this was applied to a HiTrap Ni2+ column (GE Healthcare) and eluted using an imidazole gradient concentration of 0 to 1.0 M and a final purification step of gel filtration with a HiLoad Superdex 75 16/60 column (GE Healthcare) in 0.05 M Tris-HCl (pH 8.0) and 0.1 M NaCl (supplemental Methods).
Crystallization, data collection, and structure determination
FXIIFnII was mixed with gC1qR and 50 μM ZnCl2, and the complex was isolated using a Superdex increase 200 10/300 column equilibrated with 0.02 M N-2-hydroxyethylpiperazine-N′-2-ethanesulfonic acid (pH 7.4) and 0.14 M NaCl (Figure 1C). FXIIFnII-gC1qR complex fractions were collected and concentrated to 5.3 mg/mL. Crystals were obtained in 0.1 M NaCacodylate (pH 5.5), 0.1 M CaAc2, and 12% (w/v) PEG 8000 at 10°C. A data set for a single FXIIFnII-gC1qR complex crystal was collected and processed using the CCP4 suite and the structure determined by molecular replacement with Phaser25 and the gC1qR structure (Protein Data Bank [PDB] accession number 1P32) as a template. Model building (COOT) and refinement (REFMAC)26 provided a final refined model containing the gC1qR trimer, FXIIFnII, and 3 Zn2+ (Table 1).
Crystallographic data collection and refinement statistics
| FXIIFnII-gC1qR | |
|---|---|
| Data collection | |
| Space group | I121 |
| Cell dimensions | |
| a, b, c (Å) | 106.3, 71.6, 115.9 |
| α, β, γ (°) | 90, 110.6, 90 |
| Resolution (Å) | 91.0-3.1 |
| Rmerge (%)* | 10.9 (45.5)† |
| I / σI | 7.1 (2.0)† |
| Completeness (%) | 99.9 (100.0)† |
| Redundancy | 3.1 (3.1)† |
| Wavelength (Å) | 0.97949 |
| Refinement | |
| No. of reflections | 14 378 |
| Rwork‡/Rfree (%) | 0.192/0.250 |
| No. of atoms | |
| Protein | 4752 |
| Zn2+ | 3 |
| Water | 10 |
| B-factors (Å2) | |
| Protein | 76.8 |
| Metal | 72.1 |
| Water | 46.7 |
| Root-mean-square deviations | |
| Bond lengths (Å) | 0.012 |
| Bond angles (°) | 1.57 |
| FXIIFnII-gC1qR | |
|---|---|
| Data collection | |
| Space group | I121 |
| Cell dimensions | |
| a, b, c (Å) | 106.3, 71.6, 115.9 |
| α, β, γ (°) | 90, 110.6, 90 |
| Resolution (Å) | 91.0-3.1 |
| Rmerge (%)* | 10.9 (45.5)† |
| I / σI | 7.1 (2.0)† |
| Completeness (%) | 99.9 (100.0)† |
| Redundancy | 3.1 (3.1)† |
| Wavelength (Å) | 0.97949 |
| Refinement | |
| No. of reflections | 14 378 |
| Rwork‡/Rfree (%) | 0.192/0.250 |
| No. of atoms | |
| Protein | 4752 |
| Zn2+ | 3 |
| Water | 10 |
| B-factors (Å2) | |
| Protein | 76.8 |
| Metal | 72.1 |
| Water | 46.7 |
| Root-mean-square deviations | |
| Bond lengths (Å) | 0.012 |
| Bond angles (°) | 1.57 |
Rmerge = sum(h) [sum(j) [I(hj) − <Ih>]/sum(hj) <Ih>, where I is the observed intensity and <Ih> is the average intensity of multiple observations from symmetry-related reflections calculated.
Values in parentheses are for highest-resolution shell.
Rwork = sum(h) ||Fo|h − |Fc|h| / sum(h)|Fo|h, where Fo and Fc are the observed and calculated structure factors, respectively. Rfree was computed as for Rwork, but only for (5%) randomly selected reflections, which were omitted in refinement, calculated using REFMAC.
Surface plasmon resonance (SPR)
Plasma-purified FXII was purchased from Enzyme Research Laboratories and immobilized onto a CM5 chip using an Amine Coupling Kit (GE Healthcare), and experiments were performed on a Biacore 3000 instrument. The running buffer was 0.02 M N-2-hydroxyethylpiperazine-N′-2-ethanesulfonic acid (pH 7.4), 0.14 M NaCl, and 50 μΜ ZnCl2, and the chip surface was regenerated with 1M NaCl and 0.02 M EDTA. To assess any nonspecific binding, the analyte (gC1qR) was also injected over an empty flow cell. Binding curves were analyzed on the basis of the SPR response units recorded at equilibrium for each analyte protein concentration, and a Hill plot was generated using Prism 6 (GraphPad Software).
Isothermal titration calorimetry (ITC)
A MicroCal VP ITC system (Malvern) was used for all ITC experiments performed at 25°C. The ITC cell contained 1.4 mL of wt-gC1qR or gC1qR variant while 300 μL of D5 ligand was loaded into the syringe. The reference power was set at 5 μcal/s, and the syringe stirring speed was set at 300 rpm. An initial pre-equilibration step of 1 hour was followed by 30 × 10 μL injections. Ligand dilution effects were tested by running a ligand to buffer control using the same titration parameters. The ligand to buffer control was subtracted from the experimental data, and any anomalous titration points were removed. Curves for D5-2 and HK 493-516 were fit to a 1-binding-site model, whereas curves for D5 and D5-1 were fit to a 3-site sequential binding model (Table 2).
ITC-binding measurements of the HKD5 interaction with gC1qR
| Binding model | N | KD (nM) | ΔH (kJ mol−1) | ΔS (J K−1 M−1) | ΔG (kJ mol−1) | |
|---|---|---|---|---|---|---|
| D5 | 3-site sequential binding | — | 1) 1.9 (±0.1) | 1) −268.8 (±1.1) | 1) −732.7 | 1) −50.5 |
| 2) 64.9 (±1.9) | 2) −105.2 (±1.3) | 2) −214.8 | 2) −41.2 | |||
| 3) 1011.1 (±75.8) | 3) −23.5 (±1.4) | 3) 36.0 | 3) −34.3 | |||
| D5-1 | 3-site sequential binding | — | 1) 73.0 (±4.0) | 1) −200.9 (±1.4) | 1) −535.9 | 1) −41.2 |
| 2) 1579.8 (±145.3) | 2) −92.2 (±4.9) | 2) −198.5 | 2) −33.0 | |||
| 3) 2673.8 (±257.7) | 3) −62.2 (±5.9) | 3) −101.7 | 3) −31.9 | |||
| D5-2 | Single site | 2.30 (±0.01) | 763.4 (±27.5) | −1459.1 (±1.1) | −382.7 | −35.1 |
| HK 496-516 | Single site | 2.56 (±0.02) | 1612.9 (±90.3) | −164.0 (±1.8) | −439.6 | −33.2 |
| Binding model | N | KD (nM) | ΔH (kJ mol−1) | ΔS (J K−1 M−1) | ΔG (kJ mol−1) | |
|---|---|---|---|---|---|---|
| D5 | 3-site sequential binding | — | 1) 1.9 (±0.1) | 1) −268.8 (±1.1) | 1) −732.7 | 1) −50.5 |
| 2) 64.9 (±1.9) | 2) −105.2 (±1.3) | 2) −214.8 | 2) −41.2 | |||
| 3) 1011.1 (±75.8) | 3) −23.5 (±1.4) | 3) 36.0 | 3) −34.3 | |||
| D5-1 | 3-site sequential binding | — | 1) 73.0 (±4.0) | 1) −200.9 (±1.4) | 1) −535.9 | 1) −41.2 |
| 2) 1579.8 (±145.3) | 2) −92.2 (±4.9) | 2) −198.5 | 2) −33.0 | |||
| 3) 2673.8 (±257.7) | 3) −62.2 (±5.9) | 3) −101.7 | 3) −31.9 | |||
| D5-2 | Single site | 2.30 (±0.01) | 763.4 (±27.5) | −1459.1 (±1.1) | −382.7 | −35.1 |
| HK 496-516 | Single site | 2.56 (±0.02) | 1612.9 (±90.3) | −164.0 (±1.8) | −439.6 | −33.2 |
| gC1qR construct | D5 construct | N | KD1 (nM) | KD2 (nM) | KD3 (nM) | |
|---|---|---|---|---|---|---|
| WT | D5 | — | 1.9 (±0.1) | 64.9 (±1.9) | 1011.1 (±75.8) | |
| G2-5Ala | D5 | — | 1.2 (±0.1) | 33.4 (±5.7) | 724.6 (±178.3) | |
| DelG1 | D5 | — | 1.5 (±0.2) | 44.8 (±6.0) | 1283.7 (±238.8) | |
| G1-5Ala | D5 | — | 3.4 (±0.4) | 120.3 (±11.5) | 1083.4 (±176.6) | |
| DelG3 | D5 | No fit | ||||
| WT | D5-1 | — | 73.0 (±4.0) | 1579.8 (±145.3) | 2673.8 (±257.7) | |
| G2-5Ala | D5-1 | — | 131.1 (±43.0) | 1084.6 (±517.4) | 3663.0 (±369.1) | |
| DelG1 | D5-1 | — | 135.8 (±37.3) | 1228.5 (±407.9) | 3690.0 (±638.4) | |
| G1-5Ala | D5-1 | — | 120.8 (±55.4) | 680.3 (±420.0) | 1557.3 (±369.1) | |
| DelG3 | D5-1 | No fit | ||||
| WT | D5-2 | 2.3 (±0.01) | 763.4 (±27.5) | |||
| G2-5Ala | D5-2 | 2.1 (±0.02) | 885.0 (±31.9) | |||
| DelG1 | D5-2 | 2.3 (±0.03) | 724.6 (±50.0) | |||
| G1-5Ala | D5-2 | 2.3 (±0.02) | 800.0 (±45.6) | |||
| DelG3 | D5-2 | 0.5 (±0.02) | 2032.5 (±233.7) | |||
| gC1qR construct | D5 construct | N | KD1 (nM) | KD2 (nM) | KD3 (nM) | |
|---|---|---|---|---|---|---|
| WT | D5 | — | 1.9 (±0.1) | 64.9 (±1.9) | 1011.1 (±75.8) | |
| G2-5Ala | D5 | — | 1.2 (±0.1) | 33.4 (±5.7) | 724.6 (±178.3) | |
| DelG1 | D5 | — | 1.5 (±0.2) | 44.8 (±6.0) | 1283.7 (±238.8) | |
| G1-5Ala | D5 | — | 3.4 (±0.4) | 120.3 (±11.5) | 1083.4 (±176.6) | |
| DelG3 | D5 | No fit | ||||
| WT | D5-1 | — | 73.0 (±4.0) | 1579.8 (±145.3) | 2673.8 (±257.7) | |
| G2-5Ala | D5-1 | — | 131.1 (±43.0) | 1084.6 (±517.4) | 3663.0 (±369.1) | |
| DelG1 | D5-1 | — | 135.8 (±37.3) | 1228.5 (±407.9) | 3690.0 (±638.4) | |
| G1-5Ala | D5-1 | — | 120.8 (±55.4) | 680.3 (±420.0) | 1557.3 (±369.1) | |
| DelG3 | D5-1 | No fit | ||||
| WT | D5-2 | 2.3 (±0.01) | 763.4 (±27.5) | |||
| G2-5Ala | D5-2 | 2.1 (±0.02) | 885.0 (±31.9) | |||
| DelG1 | D5-2 | 2.3 (±0.03) | 724.6 (±50.0) | |||
| G1-5Ala | D5-2 | 2.3 (±0.02) | 800.0 (±45.6) | |||
| DelG3 | D5-2 | 0.5 (±0.02) | 2032.5 (±233.7) | |||
ITC-derived thermodynamic properties for the binding of HKD5, D5-1 and D5-2 with gC1qR, and individual KD values for the binding of HKD5 to the wt-gC1qR and gC1qR variant proteins. N values are not provided for the 3-site sequential binding fits, as the stoichiometry is fixed at 3. Values in parentheses is the error calculated based on the ITC curve fitting.
Plasma-based coagulation assays
Blood was drawn from healthy volunteers into vacuettes (Greiner Bio-One) containing 3.2% sodium citrate. To isolate platelet poor plasma, tubes were spun at 1860g for 30 minutes at 4°C. Pooled normal plasma (PNP) was derived from ≥20 donors, portioned in aliquots, and stored at −70°C. Ethical approval was obtained from the University of Aberdeen College Ethics Review Board. The activated partial thromboplastin time assay was performed in a STart 4 coagulometer (Diagnostica Stago). PNP ± gC1qR (50-200 µg/mL) was incubated with Zn2+ (50 µM) at 37°C for 180 seconds. Clotting was initiated by addition of CaCl2 (0.0083 M). Thrombin generation was measured in Calibrated Automated Thrombinoscope. PNP or FXII- or FXI-deficient plasma (Hypen Biomed) ± gC1qR (50-200 µg/mL) and Zn2+ (50 µM) was added to thrombin calibrator and MP reagent (Stago United Kingdom) in Greiner 96-well plates. The plate was incubated for 10 minutes followed by addition of FluCa solution, as per the manufacturer’s guidelines. Thrombinoscope software package (Synapse Bv) was used to quantify real-time thrombin activity.
Results
Structure of the FXIIFnII-gC1qR complex
Recombinant FXIIFnII domain (residues 1-71) was produced in insect cells and shown using gel filtration to bind gC1qR in the presence of 50 μM ZnCl2 (Figure 1B-C). Complex formation was not observed at lower ZnCl2 concentrations, consistent with the previous report by Joseph et al on the Zn2+ dependency of FXII binding to gC1qR.7 The FXIIFnII-gC1qR structure was determined to 3Å resolution (Table 1 and Figure 1D). Figure 1E shows FXIIFnII has an asymmetric binding mode and unexpected stoichiometry of 1 FXIIFnII bound to the gC1qR trimer. The asymmetry of binding is achieved in part by the Arg36 and Arg65 side chains of FXII reaching out via the guanidinium groups to form contacts with distinct gC1qR negatively charged pockets termed G1 and G2 (supplemental Videos 1 and 2). gC1qR pocket G1 consists of anionic residues 190 to 202 (termed the G1-loop) together with residues Tyr236 from the αB helix and Trp233, Asp229 from the αB-β7 loop burying a surface area of 489 Å2. The FXIIFnII β2 strand forms main-chain to main-chain interactions resulting in an antiparallel β-sheet contact with gC1qR residues Glu198-Ser201 (Figure 2A-B). Electrostatic complementarity occurs via negatively charged side chains from gC1qR (Asp197, Asp229) forming salt bridge interactions with FXII side chains Lys45 and Arg36, respectively. At the tip of the FXIIFnII β1-β2 hairpin, the Arg36 side chain forms further hydrogen bonds to the main-chain carbonyl of Thr228 and a cation-π interaction with the side chain of Tyr236. Central hydrophobic contacts are made by the side chain of FXII Tyr39 with gC1qR Ala199 and flanking this FXII Gln37 hydrogen bonds to the side chain of gC1qR Ser201 (Figure 2B).
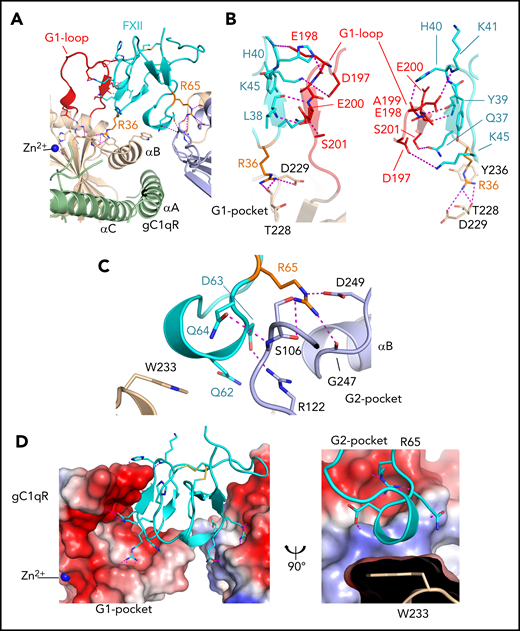
Structure of the FXIIFnII domain gC1qR interface. (A) Diagram of the FXIIFnII-gC1qR complex colored as in Figure 1E. The gC1qR G1-loop, residues 196 to 204 (red), and pockets G1 and G2 are labeled interacting with FXII residues Arg36 and Arg65 (orange) shown as sticks. Electrostatic interactions are shown as purple dotted lines and the Zn2+ as a blue sphere. (B) Interactions of the gC1qR G1-loop with FXII are shown as 2 different views. (C) The gC1qR G2 pocket is shown (purple), and key interacting FXII residues and the gC1qR residue Trp233 are shown as sticks (wheat). (D) A charged surface representation of the gC1qR G1 and G2 pockets, with FXIIFnII in cyan as a cartoon diagram with key interacting side chains as sticks.
Structure of the FXIIFnII domain gC1qR interface. (A) Diagram of the FXIIFnII-gC1qR complex colored as in Figure 1E. The gC1qR G1-loop, residues 196 to 204 (red), and pockets G1 and G2 are labeled interacting with FXII residues Arg36 and Arg65 (orange) shown as sticks. Electrostatic interactions are shown as purple dotted lines and the Zn2+ as a blue sphere. (B) Interactions of the gC1qR G1-loop with FXII are shown as 2 different views. (C) The gC1qR G2 pocket is shown (purple), and key interacting FXII residues and the gC1qR residue Trp233 are shown as sticks (wheat). (D) A charged surface representation of the gC1qR G1 and G2 pockets, with FXIIFnII in cyan as a cartoon diagram with key interacting side chains as sticks.
The FXII Arg65 side chain utilizes the guanidinium group to form interactions with 3 gC1qR residues: a salt bridge to Asp249, a hydrogen bond to the Gly247 main-chain carbonyl, and hydrogen bonds to the Ser106 main and side chain (labeled G2 pocket in Figure 2C). Additional gC1qR G2 pocket interactions occur, with FXII residues Asp63 and Gln64 forming a salt bridge and hydrogen bond to the gC1qR Arg122 side chain and Ser106 main-chain nitrogen, respectively, burying a total surface area of 238 A2. Between the G1 and G2 pockets, a hydrophobic contact is formed between the FXIIFnII Gln62 and the gC1qR Trp233 side chain (Figure 2D).
Quantitation of FXII-gC1qR ligand binding
The FXIIFnII-gC1qR structure is consistent with previous enzyme-linked immunosorbent assay data from Gebrehiwet et al showing the single point mutation of gC1qR Trp233Gly and G1-loop deletion variants disrupted FXII binding.27 The involvement of the gC1qR G2 pocket residues in the interaction with FXII is novel, and we developed an SPR assay to quantitate the gC1qR-FXII interaction with a series of gC1qR variants. Plasma-purified full-length FXII was amine coupled to a CM5 sensor chip (GE Healthcare), and a reference cell was prepared by blank amine coupling. A recombinant gC1qR wild-type (WT-gC1qR) dilution series was conducted, and analysis of sensorgrams resulted in an equilibrium dissociation constant (KD) of 120 ± 12 nM in the presence of 50 μM ZnCl2 (Figure 3a). Consistent with the gel filtration data, no complex was observed in the absence of Zn2+ or with EDTA in excess (5 mM). To test the contribution of the gC1qR G1 and G2 pockets to binding FXII, we prepared 2 gC1qR variants replacing key residues with alanine: (1) T228A, D229A, W233A, and Y236A (variant gC1qR-G1-4Ala) and (2) S106A and D249A (gC1qR-G2-2Ala). The structural integrity of the recombinant gC1qR variants was confirmed by gel filtration, revealing native-like characteristics of the trimer. Both the gC1qR G1 and G2 pocket variants failed to elicit a significant signal response or a clear association binding curve under the same conditions of the SPR binding assay to FXII, illustrating the contribution of residues from the G1 and G2 pocket to FXII binding (Figure 3B-C).
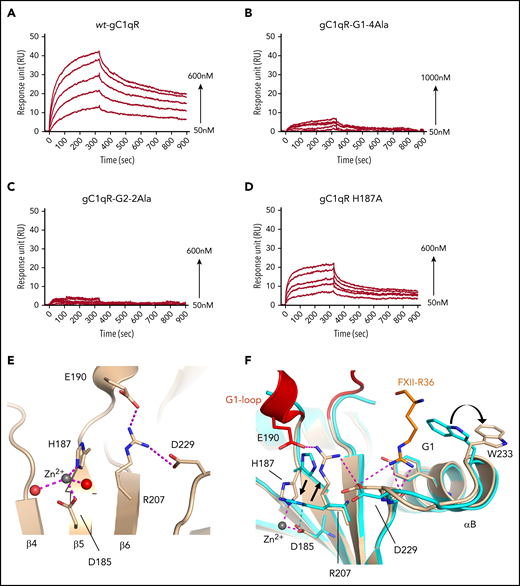
Quantitation of gC1qR binding to FXII using SPR. Plots of SPR sensorgrams measured in response units on the y-axis are shown illustrating wt-gC1qR (A) and gC1qR variants (B-D) binding to immobilized full-length FXII at increasing concentrations indicated. (B) gC1qR variant with 4 Ala substitutions made in the region of the G1 pocket (T228A, D229A, W233A, and Y236A). (C) gC1qR G2-pocket variant (S106A and D249A) and (D) gC1qR variant H187A (Zn2+-binding site ablation). (E) Cartoon diagram showing a close-up view of the gC1qR Zn2+-binding site, with key residues shown as sticks and electrostatic interactions shown as dashed lines (purple). The Zn2+ (gray) and water molecules (red) are shown as solid spheres. (F) Superposition of the FXIIFnII-bound gC1qR (colored wheat with the FXII side chain Arg36 in orange) with the unbound gC1qR structure (cyan) in the region of the G1 pocket illustrating conformational changes. The Arg207 side chain replaces the Zn2+ in coordinating the Asp185 side chain in the ligand-free gC1qR structure. Black arrows indicate gC1qR side-chain movements from ligand-free to FXIIFnII bound. Electrostatic interactions are shown as purple dotted lines.
Quantitation of gC1qR binding to FXII using SPR. Plots of SPR sensorgrams measured in response units on the y-axis are shown illustrating wt-gC1qR (A) and gC1qR variants (B-D) binding to immobilized full-length FXII at increasing concentrations indicated. (B) gC1qR variant with 4 Ala substitutions made in the region of the G1 pocket (T228A, D229A, W233A, and Y236A). (C) gC1qR G2-pocket variant (S106A and D249A) and (D) gC1qR variant H187A (Zn2+-binding site ablation). (E) Cartoon diagram showing a close-up view of the gC1qR Zn2+-binding site, with key residues shown as sticks and electrostatic interactions shown as dashed lines (purple). The Zn2+ (gray) and water molecules (red) are shown as solid spheres. (F) Superposition of the FXIIFnII-bound gC1qR (colored wheat with the FXII side chain Arg36 in orange) with the unbound gC1qR structure (cyan) in the region of the G1 pocket illustrating conformational changes. The Arg207 side chain replaces the Zn2+ in coordinating the Asp185 side chain in the ligand-free gC1qR structure. Black arrows indicate gC1qR side-chain movements from ligand-free to FXIIFnII bound. Electrostatic interactions are shown as purple dotted lines.
Zn2+ binding to the gC1qR receptor induces a conformational change
Kumar et al showed gC1qR displays specific binding to Zn2+ ions,28 and divalent cations such as Ca2+ and Mg2+ do not bind or support formation of the complex with FXII. Utilizing the structure factors collected for the FXIIFnII-gC1qR complex, we observed the 3 highest peaks in an anomalous difference Fourier map were consistent with a Zn2+ coordinated by residues Asp185 and His187. The measured bond lengths are consistent with Zn2+ tetrahedral coordination geometry with 2 water molecules also bound. To test the contribution of the gC1qR Zn2+-binding site to FXII binding, we prepared a gC1qR H187A variant. SPR experiments were used to assess binding to immobilized full-length FXII and revealed attenuated binding of gC1qR H187A compared with wt-gC1qR, which could not be fitted to derive a KD (Figure 3D). The Zn2+ is located close to the base of the acidic G1-loop adjacent to the FXIIFnII-binding site (Figure 3E). In the previously published gC1qR crystal structure (PDB accession number 1P3216 ), no metal ion is observed bound at the Asp185-His187 site. We also determined a 1.7-Å-resolution gC1qR structure in the presence of Ca2+ ions, and as expected, no metal ion was bound between residues His187 and Asp185 (unpublished results).
A comparison of the ligand-free gC1qR structures with the FXII-gC1qR complex reveals that the Zn2+ is replaced by the Arg207 side chain, which forms a salt bridge with Asp185 (Figure 3F). Arg207 does not directly contact FXII, but in the FXII-gC1qR complex, it forms a salt bridge to Glu190, which could indirectly influence FXII binding via the G1-loop. The Trp233 side chain is observed to have multiple conformations in the unbound crystal structures and resembles a flexible tip of the thumb-like helix αB, which can open and close to allow ligand access to the G1 pocket (supplemental Video 3). The other major difference is that the anionic G1-loop becomes ordered upon FXIIFnII binding, and this loop is not resolved in the unbound gC1qR crystal structures and is assumed to be flexible. These 2 sets of conformational changes in the region of the G1 pocket suggest an indirect/allosteric basis for the Zn2+ modulation of FXII binding.
HKD5 binding to gC1qR
HK binding to gC1qR has been quantified by a number of different techniques,7,29-32 and SPR data in the study published by Pixley et al revealed that binding to full-length HK was in the range of 0.7 to 0.8 nM, which could be abolished in the presence of chelating agent EDTA.33 The HKD5 domain and constituent peptides have been characterized as the key cell and Zn2+-binding34-36 sites of HK. To build on the previous data, conduct fine mapping of HKD5-binding regions, and determine the stoichiometry of the interaction with the gC1qR trimer, we expressed a series of HKD5 constructs (Figure 4A). Using gel filtration, we were able to detect the coelution of the HKD5-gC1qR complex when applied mixed in a 2:1 ratio, whereas a 3:1 ratio revealed excess unbound HKD5 eluting separately (Figure 4B). To explore this further, we used ITC with gC1qR in the sample cell titrated with HKD5. These data confirmed that HKD5 binds to gC1qR in the absence of Zn2+, and binding was unaffected by an excess of EDTA in the ligand buffer (Figure 4C). Examination of the binding isotherm revealed a multistep binding curve that could be modeled as 3 sequential binding steps with very different affinities (KD values: ∼1.9 nM, ∼64.9 nM, and ∼1.01 μM). The symmetrical gC1qR trimer is therefore unable to accommodate HKD5 at the 3 equivalent sites, demonstrating allosteric effects between sites resulting in asymmetric binding. The first event is of high affinity (KD of ∼1.9 nM) and is associated with a strongly exothermic interaction and a compensating negative entropy change, typical of the binding and immobilization of a flexible ligand (Table 2). The second and third binding events are 35- and 500-fold weaker, with much diminished enthalpy and entropy changes, consistent with partial steric occlusion at these 2 sites once the first high-affinity site is occupied.
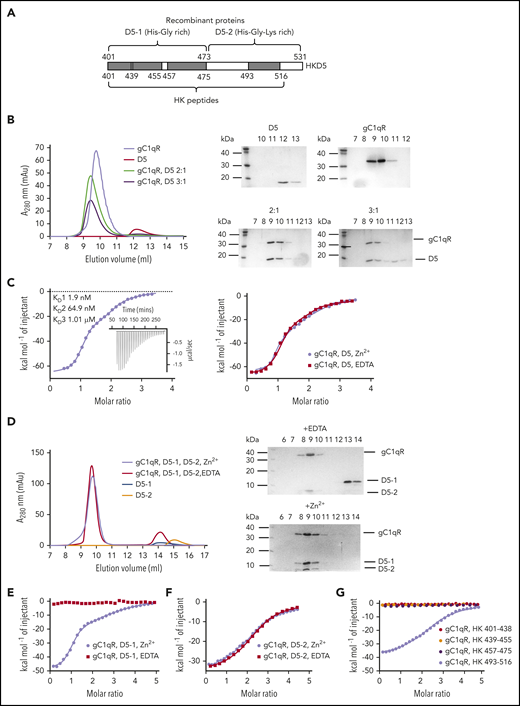
Analysis of gC1qR binding to HKD5. (A) The recombinant HKD5 construct boundaries are shown with residue numbers indicated for D5-1, D5-2, and shorter peptides used in the ITC and gel filtration experiments. (B) Gel filtration of HKD5 combined with gC1qR at different molar ratios. Elution profiles are shown on the left, and Coomassie-stained SDS-PAGE gels of the fractions collected are shown on the right. The 3:1 ratio of HKD5 to gC1qR reveals excess HKD5 eluting separately suggesting the trimer only supports 2 HKD5 polypeptides. (C) ITC measurements of gC1qR binding to HKD5. D5 was titrated into gC1qR in the presence of Zn2+ or EDTA. This was fit to a 3-site sequential binding model with no difference between curves produced in the presence or absence of Zn2+. (D) Gel filtration of D5-1, D5-2, and gC1qR in the presence of 50 μM ZnCl2 (black) or 5 mM EDTA (red) revealing D5-1 has a Zn2+ dependence whereas HKD5-2 does not. (E) ITC experiments with D5-1 and D5-2 respectively titrated into gC1qR in the presence of Zn2+. Similarly to full-length HKD5, D5-1 was fitted to a 3-site sequential binding model, and binding was Zn2+ dependent. (F) The binding of D5-2 was Zn2+-independent and was fit to a single-site binding model with a calculated N = 2. (G) HKD5 derived shorter peptides titrated into gC1qR. All titrations excluding HK 493-516 were performed in the presence of Zn2+. HK 493-516 was the only peptide to show binding, and the curve resulted in binding affinities and N values comparable to D5-2.
Analysis of gC1qR binding to HKD5. (A) The recombinant HKD5 construct boundaries are shown with residue numbers indicated for D5-1, D5-2, and shorter peptides used in the ITC and gel filtration experiments. (B) Gel filtration of HKD5 combined with gC1qR at different molar ratios. Elution profiles are shown on the left, and Coomassie-stained SDS-PAGE gels of the fractions collected are shown on the right. The 3:1 ratio of HKD5 to gC1qR reveals excess HKD5 eluting separately suggesting the trimer only supports 2 HKD5 polypeptides. (C) ITC measurements of gC1qR binding to HKD5. D5 was titrated into gC1qR in the presence of Zn2+ or EDTA. This was fit to a 3-site sequential binding model with no difference between curves produced in the presence or absence of Zn2+. (D) Gel filtration of D5-1, D5-2, and gC1qR in the presence of 50 μM ZnCl2 (black) or 5 mM EDTA (red) revealing D5-1 has a Zn2+ dependence whereas HKD5-2 does not. (E) ITC experiments with D5-1 and D5-2 respectively titrated into gC1qR in the presence of Zn2+. Similarly to full-length HKD5, D5-1 was fitted to a 3-site sequential binding model, and binding was Zn2+ dependent. (F) The binding of D5-2 was Zn2+-independent and was fit to a single-site binding model with a calculated N = 2. (G) HKD5 derived shorter peptides titrated into gC1qR. All titrations excluding HK 493-516 were performed in the presence of Zn2+. HK 493-516 was the only peptide to show binding, and the curve resulted in binding affinities and N values comparable to D5-2.
HKD5 Zn2+-dependent component to gC1qR binding
To determine the origin of the allosteric component of the interaction, we expressed recombinant N- and C-terminal fragments of HKD5 in which D5-1 (residues 401-473) contains the His-Gly-rich region and D5-2 (residues 474-531) is His-Gly-Lys rich (Figure 4a). HKD5 has a His-rich nature (21% of the HKD5 sequence) and has previously been shown to bind Zn2+ ions.34,35 Using electrospray ionization mass spectrometry with the whole HKD5 we detected polypeptide species with 1, 2, and 3 bound Zn2+ ions and 2 Zn2+ bound in each case to D5-1 and D5-2 constructs, respectively (supplemental Figures 3-5).
We examined the binding of the D5-1 and D5-2 fragments to gC1qR by gel filtration and showed that both components could bind independently. The coelution of the N-terminal D5-1 with gC1qR had a Zn2+ ion dependency, whereas D5-2 did not. Coelution of D5-1 with gC1qR could be eliminated in the presence of EDTA, but D5-2 was unaffected by EDTA (Figure 4D). This was confirmed by ITC measurements, in which titrations in buffer containing EDTA eliminated the interaction with D5-1 but had no effect on D5-2 (Figure 4E-F).
The binding isotherms of the 2 fragments also showed highly distinct ITC profiles with HKD5-1 retaining the complex 3-phase binding curve observed for full-length HKD5, whereas the C-terminal D5-2 fragment could be modeled as a single-phase binding event, with an estimated stoichiometry of between 2 and 3 (N = 2.3) gC1qR sites each with a KD of ∼763.4 nM. The 3-phase interaction of D5-1 with gC1qR showed a >40-fold reduction in affinity for the first high-affinity step compared with full-length HKD5 but only a 25 to threefold reduction for the lower affinity steps 2 and 3. These data show that the allosteric effects are largely associated with the His-Gly-rich D5-1 motif and occur in a Zn2+-dependent manner (Table 2). The observation that the D5-2 fragment binds in a Zn2+-independent fashion suggests that this basic His-Gly-Lys-rich D5-2 motif may be interacting at a different location on the gC1qR structure to D5-1. In the presence of Zn2+, we were able to detect a tricomplex among gC1qR, D5-1, and D5-2 by gel filtration, with all components comigrating in the same fractions (Figure 4D).
To further delineate the site of interaction on D5, we considered still shorter peptide motifs derived from D5-1 (HK residues 401-438, HK 439-455, and HK 457-475) and D5-2 (HK 493-516) and studied the interaction by ITC. None of the peptides derived from D5-1 revealed any binding in the presence or absence of Zn2+ ions to gC1qR, suggesting that the binding site covers a much larger proportion of the D5-1 fragment (Figure 4G). However, the His-Gly-Lys-rich peptide HK 493-516 derived from D5-2 produced a Zn2+ independent single-site binding isotherm comparable to that for D5-2, differing by only twofold in binding affinity, which similarly indicated ∼3 equivalent sites on the gC1qR trimer. The HK 493-516 sequence resembles other peptides rich in Gly-Lys that have been characterized as binding to gC1qR37,38 without Zn2+, and an alignment of these sequences is shown in supplemental Figure 7C.
A central gC1qR loop is used for HKD5 binding
Ghebrehiwet et al used deletion mutants to identify gC1qR residues 144 to 148, 196 to 202, and 204 to 218 as being important for whole HK binding.27 To determine the location of the recombinant HKD5- and D5-1– and D5-2 fragment–binding sites, we prepared 4 similar gC1qR variants, removing negatively charged residues from the anionic loop regions: gC1qR-G2-5Ala (residues 146-148 and 156-157 mutated to Ala), gC1qRdelG3 (G3-loop residues 214-224 deleted), gC1qRdelG1 (G1-loop residues 196-200 deleted), and a G1-loop variant (residues 196-200), with 5 acidic residues substituted for Ala termed gC1qR-G1-5Ala (supplemental Figure 6). ITC experiments revealed only the gC1qRdelG3 variant had a significant effect in abrogating binding of HKD5 with other variants reproducing the multiphase binding isotherms evident for WT-gC1qR (Figure 5A-C). We repeated the titration with the fragments D5-1 and D5-2 and observed no detectable binding to D5-1 and significant attenuation of binding to D5-2 (Figure 5B-C). This indicates that the central acidic G3-loop that defines an inner pocket (G3 pocket) plays a significant role in the interaction with HKD5, particularly for the binding of the N-terminal D5-1 region (Figure 5D; supplemental Video 4). The placement of the G3-loop at the center of the gC1qR trimer is consistent with the allosteric nature of HKD5 and D5-1 binding, as steric occlusion would reduce the ability of subsequent HKD5 ligands to cobind (Figure 5E). Overall the anatomy of the gC1qR monomer is such that it resembles a hand with the FXII-binding site formed between the index finger (G1 loop) and the thumb (αB), and the palm of the hand contains the Zn2+-binding site, adjacent to which is the little finger (G3-loop), defining the principal HK-binding site (Figure 5D; supplemental Video 4).

Mapping of the gC1qR-binding site for HKD5. (A-C) ITC experiments with HKD5, D5-1, and D5-2, respectively titrated against the gC1qR variants compared with wt-gC1qR. Deletion of the gC1qR G3-loop (gC1qRdelG3) in blue showed no binding to D5-1 and reduced binding to both HKD5 and D5-2, whereas the other variants were comparable to wt-gC1qR. (D) The HKD5-binding site maps to the region of the G3-loop (blue) that forms the boundary of a pocket (G3, shown as blue dashed ellipse), which extends across the β-sheet to the Zn2+ site (gray sphere). The locations of the G1 pocket (purple dashed ellipse), G2-loop (orange), and G1-loop (red) are shown. (E) A schematic representation of the proposed D5 binding to gC1qR. The ligand-free gC1qR and HKD5 are shown top left. D5-1 is represented as a larger circle (blue) connected to a smaller circle representing D5-2 (red). The larger size of the D5-1 circle is representative that this region binding to gC1qR cannot be emulated by short peptides. gC1qR is shown in gray to represent the Zn2+-free form, and flexible anionic loops are shown colored as in panel D with the G1- and G2-loops radially located and the G3-loop in the center. The binding of D5-1 is sequential whereby tight binding of the first D5-1 (KD1) is followed by subsequently reduced affinity second and third binding events (KD2 and KD3), suggesting a third HKD5 binding is sterically occluded (shown as transparent). The binding of D5-2 to gC1qR is not sequential, and all binding events to gC1qR have equivalent affinity (KD1).
Mapping of the gC1qR-binding site for HKD5. (A-C) ITC experiments with HKD5, D5-1, and D5-2, respectively titrated against the gC1qR variants compared with wt-gC1qR. Deletion of the gC1qR G3-loop (gC1qRdelG3) in blue showed no binding to D5-1 and reduced binding to both HKD5 and D5-2, whereas the other variants were comparable to wt-gC1qR. (D) The HKD5-binding site maps to the region of the G3-loop (blue) that forms the boundary of a pocket (G3, shown as blue dashed ellipse), which extends across the β-sheet to the Zn2+ site (gray sphere). The locations of the G1 pocket (purple dashed ellipse), G2-loop (orange), and G1-loop (red) are shown. (E) A schematic representation of the proposed D5 binding to gC1qR. The ligand-free gC1qR and HKD5 are shown top left. D5-1 is represented as a larger circle (blue) connected to a smaller circle representing D5-2 (red). The larger size of the D5-1 circle is representative that this region binding to gC1qR cannot be emulated by short peptides. gC1qR is shown in gray to represent the Zn2+-free form, and flexible anionic loops are shown colored as in panel D with the G1- and G2-loops radially located and the G3-loop in the center. The binding of D5-1 is sequential whereby tight binding of the first D5-1 (KD1) is followed by subsequently reduced affinity second and third binding events (KD2 and KD3), suggesting a third HKD5 binding is sterically occluded (shown as transparent). The binding of D5-2 to gC1qR is not sequential, and all binding events to gC1qR have equivalent affinity (KD1).
Isolation of a gC1qR-HK-FXII ternary complex
As both HKD5 and the FXIIFnII domain exhibit asymmetric binding to the gC1qR trimer but by different mechanisms, we next tested whether the FXIIFnII and HKD5 could bind simultaneously to gC1qR. If an excess of HKD5 is present, then FXIIFnII cannot compete for binding, and this is consistent with multiple interaction sites for HKD5 identified by ITC. However, lower stoichiometric ratios of HKD5 reveal both HKD5 and FXIIFnII coeluting in fractions from the gC1qR complex peak shown in Figure 6A. We next extended these studies to a series of gel filtration experiments using the full-length plasma-purified FXII and HK in isolation and in combination with gC1qR. The molecular weights of FXII, HK, and gC1qR estimated by reducing sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE) migration are 80 kDa, 110 kDa, and 33 kDa, respectively, and equivalent gel filtration estimates are 80 kDa (FXII monomer), 200 kDa (HK dimer39 ), and 90 kDa (gC1qR trimer). A mixture of full-length FXII, HK, and gC1qR in the presence of Zn2+ resulted in comigration of all 3 proteins in a single peak corresponding to a ∼500-kDa complex (Figure 6B). The stoichiometry of the species in this peak is equivalent to a 1:2:6 ratio of FXII-HK-gC1qR shown schematically in Figure 6C. The concept of a ternary complex formed by FXII-HK-gC1qR is consistent with previous in vitro experiments by Joseph et al showing that efficient gC1qR stimulation of PK enzymatic activity requires the presence of PK, FXII, cofactor HK, and Zn2+ ions.40
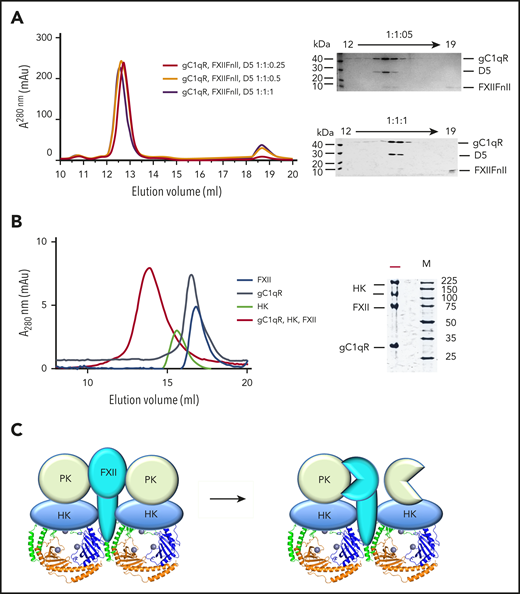
Ternary complexes of FXII, HK and gC1qR analyzed by gel filtration. (A) Analytical gel filtration elution profiles showing FXIIFnII, D5 and gC1qR combined with increasing concentrations of HKD5 in the presence of Zn2+. On the right coomassie stained SDS-PAGE gels showing gC1qR, HKD5 and FXIIFnII in the fractions collected. As the concentration of HKD5 is increased, FXIIFnII shifts from the high-molecular-weight peak to the low-molecular-weight peak, suggesting D5 is outcompeting FXIIFnII for gC1qR binding. (B) Analytical gel filtration (Superose 6 10/300) of full-length proteins HK (green), FXII (blue), gC1qR (black), and the gC1qR-HK-FXII ternary complex (red). Coomassie-stained SDS-PAGE gel showing the peak fraction of the gC1qR-HK-FXII ternary complex. (C) Schematic diagram of a hypothetical FXII-HK-gC1qR-PK complex with a 1:2:6:2 stoichiometry. In this model, gC1qR is capable of stimulating reciprocal FXII-PK activation by aligning the activation loops and active sites of the FXII and PK proteases.
Ternary complexes of FXII, HK and gC1qR analyzed by gel filtration. (A) Analytical gel filtration elution profiles showing FXIIFnII, D5 and gC1qR combined with increasing concentrations of HKD5 in the presence of Zn2+. On the right coomassie stained SDS-PAGE gels showing gC1qR, HKD5 and FXIIFnII in the fractions collected. As the concentration of HKD5 is increased, FXIIFnII shifts from the high-molecular-weight peak to the low-molecular-weight peak, suggesting D5 is outcompeting FXIIFnII for gC1qR binding. (B) Analytical gel filtration (Superose 6 10/300) of full-length proteins HK (green), FXII (blue), gC1qR (black), and the gC1qR-HK-FXII ternary complex (red). Coomassie-stained SDS-PAGE gel showing the peak fraction of the gC1qR-HK-FXII ternary complex. (C) Schematic diagram of a hypothetical FXII-HK-gC1qR-PK complex with a 1:2:6:2 stoichiometry. In this model, gC1qR is capable of stimulating reciprocal FXII-PK activation by aligning the activation loops and active sites of the FXII and PK proteases.
gC1qR effects on plasma coagulation
We next tested the effects of gC1qR on plasma coagulation in the absence of additional stimuli and observed a dose-dependent shortening of clotting time with PNP in the presence of Zn2+ (Figure 7A). Thrombin generation experiments revealed a significant shortening in the lag time on addition of gC1qR and Zn2+ to PNP, which influenced several additional parameters, including peak thrombin, endogenous thrombin potential, and velocity of thrombin generation (Figure 7B-F). The lag time of thrombin generation in the presence of gC1qR and Zn2+ was delayed by a factor of 2.8 in FXII-deficient plasma and absent in FXI-deficient plasma, indicating a dependence on the intrinsic pathway (Figure 7C). Peerschke et al also showed that gC1qR did stimulate plasma coagulation, but not in a FXII-dependent manner,41 and the reason for this discrepancy may relate to the addition of Zn2+. Analysis of 2 gC1qR variants revealed that the gC1qRdelG3 variant (but not gC1qRdelG1) was unable to stimulate thrombin generation to the same degree as wt-gC1qR, implicating the central G3-loop as being functionally important for stimulation of coagulation. It is unknown whether the concentration of endothelial cell bound gC1qR is high enough to support thrombin generation on the vessel wall.
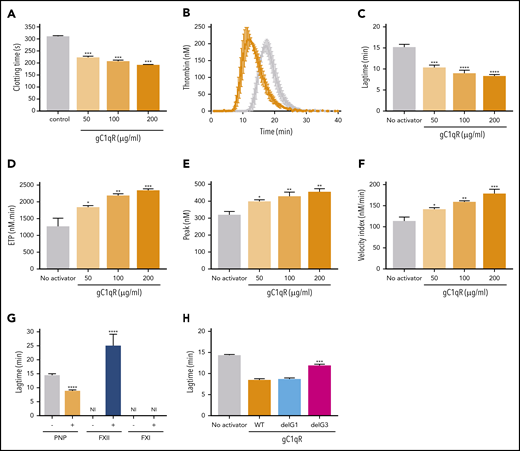
gC1qR stimulation of plasma coagulation in a FXII-dependent manner. (A) PNP was incubated for 180 seconds and increasing concentrations of gC1qR (50-200 µg/mL) with Zn2+ (50 µM). Coagulation was initiated by addition of CaCl2 (8.3 mM), and clotting time was monitored. (B-F) Thrombin generation for PNP with gC1qR (50-200 µg/mL) and Zn2+ (50 µM). Raw data curves in the presence of gC1qR (orange) and without (gray) for lag time (minutes), endogenous thrombin potential (ETP; nM/min), peak thrombin (nM), and velocity index (nM/min) were derived. (G) Lag time (minutes) is shown for thrombin generation of PNP or FXII- or FXI-deficient plasma incubated with or without gC1qR (100 µg/mL) and Zn2+ (50 µM). In the absence of gC1qR, there was no thrombin generation evident in FXI- or FXII-deficient plasma. FXII-deficient plasma with gC1qR shows a significant delay in thrombin generation, with a lag time of 25 ± 6.7 minutes vs 9 ± 0.7 minutes in PNP and no thrombin generation in FXI-deficient plasma with gC1qR, indicating a dependence on the intrinsic pathway. (H) Lag time (minutes) is shown for thrombin generation of PNP, gC1qR (100 µg/mL), and gC1qR variants with G1 (cyan) and G3-loops (pink) deleted, showing a dependency on the gC1qR G3-loop.
gC1qR stimulation of plasma coagulation in a FXII-dependent manner. (A) PNP was incubated for 180 seconds and increasing concentrations of gC1qR (50-200 µg/mL) with Zn2+ (50 µM). Coagulation was initiated by addition of CaCl2 (8.3 mM), and clotting time was monitored. (B-F) Thrombin generation for PNP with gC1qR (50-200 µg/mL) and Zn2+ (50 µM). Raw data curves in the presence of gC1qR (orange) and without (gray) for lag time (minutes), endogenous thrombin potential (ETP; nM/min), peak thrombin (nM), and velocity index (nM/min) were derived. (G) Lag time (minutes) is shown for thrombin generation of PNP or FXII- or FXI-deficient plasma incubated with or without gC1qR (100 µg/mL) and Zn2+ (50 µM). In the absence of gC1qR, there was no thrombin generation evident in FXI- or FXII-deficient plasma. FXII-deficient plasma with gC1qR shows a significant delay in thrombin generation, with a lag time of 25 ± 6.7 minutes vs 9 ± 0.7 minutes in PNP and no thrombin generation in FXI-deficient plasma with gC1qR, indicating a dependence on the intrinsic pathway. (H) Lag time (minutes) is shown for thrombin generation of PNP, gC1qR (100 µg/mL), and gC1qR variants with G1 (cyan) and G3-loops (pink) deleted, showing a dependency on the gC1qR G3-loop.
Discussion
How the contact factors assemble on the cell surface and become activated remains one of the fundamental and unanswered questions underpinning several pathways driving inflammation and thrombosis. The FXIIFnII-gC1qR-Zn2+ complex provides the first structural insight into a gC1qR ligand interaction and shows a stoichiometry of 1 FXIIFnII bound to the gC1qR trimer. The asymmetry of FXII binding to gC1qR involves 2 negatively charged surface pockets, whereas for HKD5, this is driven by the central location of a critical negatively charged loop and steric occlusion. Overall, the gC1qR ligand-binding mode we observe for FXII and HK is consistent with the molecular mechanism observed for the chaperone heat shock protein 90 (HSP90), whereby multiple client proteins can be colocalized by asymmetric binding onto the HSP90 dimer.42 The parallel with HSP90 is pertinent, as extracellular HSP90 has been described as a chaperokine43 involved in inflammatory processes and was shown to initiate PK-HK activation.44 gC1qR has a requirement for FXII, PK, and HK, whereas HSP90 requires PK and HK, but not FXII.44
FXII and HK binding to gC1qR is Zn2+ dependent, and our data show Zn2+ is bound to gC1qR residues Asp185 and His187 in between the FXII and HK-binding pockets (supplemental Video 4). These observations build on data from Kumar et al that describe a Zn2+-dependent conformational change in gC1qR28 and thus provide an allosteric mechanism for modulation of the interaction with FXII. The biological context of Zn2+ modulation of the FXII-gC1qR interaction is proposed to originate from activated endothelial cells or secreted platelet granules.45,46 How gC1qR is anchored to the cell membrane has been reported to occur via interaction with other cell receptors1 or cytokeratin-1.4 uPAR has also been shown to act as a receptor for FXII, and this interaction has been studied for endothelial,46 neutrophil,47 and dendritic cell48 function. It is unknown whether gC1qR, uPAR, and cytokeratin-mediated binding of contact factors are functionally aligned.
Gel filtration using the full-length proteins identified a FXII-HK-gC1qR complex with an overall molecular weight in the 500 kDa range, which may represent FXII bound to 2 gC1qR trimers and a HK dimer (Figure 6C). The ability of gC1qR to cluster FXII and HK into a planar ternary complex is conceptually familiar to the way vitamin K–dependent hemostatic proteases from the extrinsic pathway have their protease domains aligned with the substrate activation loop by a Ca2+-dependent process on a planar phospholipid surface.49 Targeting a chaperone to disable the function of client proteins involved in a pathogenic mechanism is established for chaperones protein disulfide isomerase50 and HSP90,42 and our data provide a scaffold for a similar approach to target gC1qR.14
The FXIIFnII-gC1qR complex crystal structure has been deposited in the PDB (www.rcsb.org; accession number 6SZW).
The online version of this article contains a data supplement.
The publication costs of this article were defrayed in part by page charge payment. Therefore, and solely to indicate this fact, this article is hereby marked “advertisement” in accordance with 18 USC section 1734.
Acknowledgments
The authors acknowledge the Diamond Light Source for provision of synchrotron radiation in using the Beamline I03.
This work was funded by the British Heart Foundation (grant RG/12/9/29775) (J.E.). Part of this work was funded by the Centre for Membrane Proteins (COMPARE) (J.E.) and an Engineering and Physical Sciences Research Council (EPSRC) studentship (A.S.).
Authorship
Contribution: B.G.K. performed the experiments with FXIIFnII and gC1qR; A.S. performed the experiments with HKD5 and gC1qR; B.G.K., A.S., I.D., and K.R.M. designed experiments, analyzed the data, and critically reviewed the manuscript; U.S. and N.J.M. performed plasma-based assays; and M.S. and J.E. contributed to the design of experiments, analysis of the data, preparation of the figures and to the writing of the manuscript.
Conflict-of-interest disclosure: J.E. receives consultant’s fees from several pharmaceutical companies. The remaining authors declare no competing financial interests.
Correspondence: Mark Searle, School of Chemistry, Biodiscovery Institute, University of Nottingham, University Park, Nottingham NG7 2RD, United Kingdom; e-mail: mark.searle@nottingham.ac.uk; and Jonas Emsley, Room C52, School of Pharmacy, Biodiscovery Institute, University of Nottingham, University Park, Nottingham NG7 2RD, United Kingdom; e-mail: jonas.emsley@nottingham.ac.uk.

This feature is available to Subscribers Only
Sign In or Create an Account Close Modal