

Key Points
Plasma MPs released during crisis in SS patients increase endothelial ICAM-1 levels and neutrophil adhesion.
HU treatment lowers PS exposure on MPs, which abolishes their proinflammatory properties.

Abstract
Microparticles (MPs) are submicron extracellular vesicles exposing phosphatidylserine (PS), detected at high concentration in the circulation of sickle cell anemia (SS) patients. Several groups studied the biological effects of MPs generated ex vivo. Here, we analyzed for the first time the impact of circulating MPs on endothelial cells (ECs) from 60 sickle cell disease (SCD) patients. MPs were collected from SCD patients and compared with MPs isolated from healthy individuals (AA). Other plasma MPs were purified from SS patients before and 2 years after the onset of hydroxyurea (HU) treatment or during a vaso-occlusive crisis and at steady-state. Compared with AA MPs, SS MPs increased EC ICAM-1 messenger RNA and protein levels, as well as neutrophil adhesion. We showed that ICAM-1 overexpression was primarily caused by MPs derived from erythrocytes, rather than from platelets, and that it was abolished by MP PS capping using annexin V. MPs from SS patients treated with HU were less efficient to induce a proinflammatory phenotype in ECs compared with MPs collected before therapy. In contrast, MPs released during crisis increased ICAM-1 and neutrophil adhesion levels, in a PS-dependent manner, compared with MPs collected at steady-state. Furthermore, neutrophil adhesion was abolished by a blocking anti–ICAM-1 antibody. Our study provides evidence that MPs play a key role in SCD pathophysiology by triggering a proinflammatory phenotype of ECs. We also uncover a new mode of action for HU and identify potential therapeutics: annexin V and anti–ICAM-1 antibodies.
Introduction
Sickle cell disease (SCD) is a genetic disorder in which the normal hemoglobin A is replaced by the abnormal hemoglobin S. More than 80% of patients with SCD have the homozygous form SS or a less frequent and less severe form (SC).1,2 Vaso-occlusive crisis (VOC) is the most common and 1 of the most severe clinical events in SCD. Intravital microscopy in SCD mouse models revealed that neutrophils play a key role in this complication,3,4 because these large cells adhere to the endothelium of postcapillary venules5 and subsequently cause trapping of sickle red blood cells (RBCs). Adhesion molecules expressed by the endothelium are crucial for neutrophil recruitment6 : P-selectin and E-selectin are involved in the rolling process, whereas ICAM-1 allows firm adhesion of these cells.7 In SCD, stimuli that cause overexpression of these molecules could trigger acute VOCs.
As in several thrombotic diseases,8 high plasma concentrations of microparticles (MPs), defined as phosphatidylserine (PS)+ bud membrane vesicles of 100 nm to 1 µm in diameter, were reported in SS patients,9-12 with a further increase during VOCs.13,14 Moreover, RBC-derived MP (RBC-MP) levels were associated with markers of hemolysis in SCD patients.15,16 Because of their exposed PS, their nucleic acids, and their enclosed or transmembrane proteins, MPs are presumed to be involved in the cellular cross talk of different processes crucial to SCD pathophysiology. Indeed, MPs released by in vitro–stimulated platelets, erythrocytes, or monocytes from non-SCD subjects were shown to modulate vascular tone, coagulation, inflammation, and adhesion of circulating cells to the endothelium.17 For example, PS and tissue factor (CD142) are exposed elements of MPs that are reported to activate the coagulation cascade.18,19 Some reports also suggest that MPs are biomarkers of several SCD complications.12,20,21 With regard to the biological effects of MPs in vivo, Vats et al showed that platelet extracellular vesicles trigger lung injury in SCD mice,22 and Camus et al showed that RBC-MPs trigger kidney vaso-occlusions via enhanced endothelial adhesion of RBCs in a murine model of SCD.23 However, the first study used platelet-derived vesicles produced in SCD mice treated with lipopolysaccharide, and the second study was performed using MPs of a unique and minor subtype that were generated ex vivo, whereas circulating MPs in SCD plasma derive from several blood cell types.11
MP composition is known to vary according to the cell type of origin and the stimuli used for their generation.24 For example, Wang et al showed that MPs generated ex vivo from the monocytic cell line THP-1 and stimulated or not with lipopolysaccharide had a 1000-fold difference in their interleukin-1β protein content.25 We also reported that, during crisis, MP concentration and externalization of PS were increased in SS patients,14 in contrast with the gradual, but intense, decrease in MP PS exposure observed during hydroxyurea (HU) treatment.26 Altogether, these observations emphasized the need to analyze the functional properties of MPs produced under different clinical conditions.
The aim of the present study was to determine the impact of MPs isolated from fresh AA (healthy donors), SC, and SS plasma on endothelial cell (EC) phenotype, focusing on neutrophil adhesion. We used in vivo–generated MPs isolated from SCD patients in different clinical conditions (ie, at steady-state, during crisis, or during HU treatment), as well as MPs isolated from AA individuals. We assessed changes at the RNA, protein, and functional levels. We report exacerbated proinflammatory properties of SS MPs obtained during crisis and uncover a new mechanism of action by which HU abolishes these properties.
Patients and methods
Patients and plasma MP purification
Sixty SCD patients regularly followed by the sickle cell center at the University Hospital of Pointe-à-Pitre (Guadeloupe) were enrolled through 3 protocols: (1) 12 SC and 17 SS patients at steady-state (supplemental Table 1; available on the Blood Web site), (2) 15 SS patients for whom samples were obtained at steady-state, as well as before HU treatment onset (HU0 MPs) and 2 years after HU treatment onset (HU24 MPs) (supplemental Table 2); and (3) 16 SS patients whose blood was sampled at steady-state (steady-state MPs) and during painful VOCs (crisis MPs) (supplemental Table 3). The last 2 protocols were designed longitudinally. Fourteen healthy Guadeloupean subjects were also enrolled. All donors were informed about the purpose and procedures of the study and gave their written informed consent. All studies were conducted in accordance with the Declaration of Helsinki and approved by the Regional Ethics Committee (Comité de Protection des Personnes Sud/Ouest Outre Mer III, Bordeaux, France; registration numbers 2009-A00211-56 and 2012‐A00701‐42 and University Hospital of Pointe-à-Pitre, Délégation à la Recherche Clinique et à l'Innovation–Centre Hospitatier Universitaire de Pointe-à-Pitre, 230214, Guadeloupe). For each donor, MPs were purified as previously described.11 Crisis MPs were purified as previously described,14 once a diagnosis of VOC (a musculoskeletal and/or visceral pain only explained by sickle cell anemia and requiring hospitalization) was made, within the first hour of Emergency Department admission and before any medication administration. Briefly, blood was collected into a 3.2% trisodium citrate tube. As a result of the overlap in size between the smallest platelets and the largest MPs,27 we did not carry out a double 2500g centrifugation, because we show that such centrifugations cause the loss of ∼67% of detected RBC-MPs (supplemental Figure 1). We instead centrifuged blood samples for 10 minutes at 1000g to obtain platelet-depleted plasma and then ultracentrifuged the obtained plasma (20 minutes at 20 817g) to pellet MPs. Two washing steps were used: the first in an EDTA-containing buffer (10 mM HEPES, 5 mM KCl, 1 mM MgCl2, 136 mM NaCl, 20 mM EDTA; pH = 7.4) to separate MPs from bound proteins or cells and the second in an EDTA-free buffer (same composition as the preceding buffer but without EDTA). Purified MPs were stored in the latter buffer at −80°C until use.
MP characterization and incubation
The cell type of origin, size, and concentration of MPs were determined by flow cytometry (FC 500; Beckman Coulter, Brea, CA), using cell type marker-specific antibodies, forward scatter signal, and calibrated fluorescent microbeads, respectively (Flow-Count; Beckman Coulter). Vesicles were considered MPs if their forward scatter and side scatter values were smaller than or equal to those of 0.9-µm polystyrene microbeads (Megamix; Biocytex, Marseille, France) and if they were positively stained with fluorescein isothiocyanate-conjugated annexin V (AV; Beckman Coulter). Staining and analysis of MPs were carried out in a Ca2+-containing buffer (10 mM HEPES, 3 mM CaCl2; pH = 7.4). When selected MPs were needed, we negatively selected MPs using a depletion kit composed of Anti-PE MicroBeads and MACS LD columns (order numbers 130-097-054 and 130-042-901; Miltenyi Biotec, Bergisch Gladbach, Germany), according to the manufacturer’s instructions. This method had already been used by another group.28 Briefly, we incubated the high dose of MPs with relevant phycoerythrin (PE)-conjugated antibodies targeting unwanted MPs (anti-CD15, anti-CD14, and anti-CD106, as well as anti-CD41 antibodies to select RBC-MPs or anti-CD235a antibodies to select platelet-derived MPs [PLT-MPs]). Then, microbeads coupled with antibody directed against PE were added to bind to these antibodies, and subsequent depletion of unwanted MPs was achieved by applying a strong magnetic field. Characterization of MPs before and after selection is shown in supplemental Figure 2.
Unless specified otherwise, the dose of MPs that we used for EC stimulation was 7 MPs per cell (also called high dose), which corresponds to a medium MP concentration ∼6 times lower than in the blood of SS patients. This high dose represents 2800 MPs per microliter for experiments in 8-well slides and 2450 MPs per microliter for experiments in 24-well plates. For experiments with selected MPs, we incubated ECs with the quantity of PLT-MPs or RBC-MPs contained in the high dose of total MPs. When appropriate, MPs were preincubated with AV (2.5 ng/µL; cat. no. 640901; BioLegend, London, United Kingdom) in the Ca2+-containing buffer or with lactadherin (2.5 ng/µL; 2767-MF-050; R&D Systems, Minneapolis, MN). We used ECs of the TrHBMEC cell line, which were the kind gift of B. Weksler.29 These ECs were cultured as described previously,30 at passage number 20 or 21. Primary human microvascular pulmonary ECs (cat. no. C-12281; PromoCell, Heidelberg, Germany) were also used, but at passage number 5 or 6. All ECs were grown to 90% confluence and then incubated without MPs (negative control) or with MPs for 4 hours, unless specified otherwise.
Determination of MPs impact by reverse-transcription quantitative polymerase chain reaction
After incubation, total RNA was extracted using an RNeasy Plus Micro Kit (cat. no. 74034; QIAGEN, Hilden, Germany) and reverse transcribed using a High-Capacity cDNA Reverse Transcription Kit (cat. no. 4368814; ThermoFisher, Saint-Aubin, France). Both steps were performed according to the manufacturers’ instructions. Complementary DNA was then analyzed by quantitative polymerase chain reaction using SYBR Select Master Mix (4472903) and a StepOne Plus thermocycler (both from ThermoFisher). TBP was used as reference gene. The primers used for quantitative polymerase chain reaction are described in supplemental Table 4.
Quantification of protein levels
After the incubation period, ECs were trypsinized and labeled with fluorescent antibodies targeting ICAM-1 (CD54, HA58), VCAM-1 (CD106, 51-10 C9; Beckman Coulter), or E-selectin (CD62E, 68-5H11). The following isotype-control antibodies were used: PE IgG1 (679.1Mc7; Beckman Coulter) and PE-Cy5 IgG1 κ (MOPC-21). If not specified, antibodies were from Becton Dickinson. Mean fluorescence intensity (MFI) and the percentage of positive cells were evaluated using flow cytometry. For enzyme-linked immunosorbent assay experiments, we used a kit for soluble ICAM-1 (sICAM-1) expression (Reference: 851590015; Eurobio, Les Ulis, France), according to the manufacturer’s instructions.
Adhesion experiments
For adhesion experiments, cells were cultured in 8-well chambers mounted on glass slides (Lab-Tek II CC2 Chamber Slide System; Life Technologies) with 100 000 ECs per well. During incubation of ECs with MPs, SCD neutrophils were isolated from blood collected into a 3.2% trisodium citrate tube and carefully added on top of a Percoll gradient with 2 densities: 1.079 over 1.098. Two consecutive centrifugations with no brake, the first at 150g for 8 minutes and the second at 1200g for 10 minutes, allowed separation of neutrophils from plasma and other cells. Contaminating erythrocytes were lysed using lysis buffer (155 mM NH4Cl, 1.0 mM KHCO3, 10 nM EDTA in distilled H2O; pH = 7.4). When appropriate, 30 minutes before incubating SCD neutrophils, a blocking anti–ICAM-1 antibody (CD54, 84H10; Sigma-Aldrich Merck, Darmstadt, Germany) was added (2.5 ng/µL) to the cell culture supernatant. At the end of the incubation period, with or without MPs (control well), fluorescently labeled neutrophils (Alexa Fluor 488 anti-CD16 antibody; 3G8; Ozyme, Saint-Cyr-L’École, France) were incubated for 30 minutes with ECs. Before removing wells and adding the cover slip, ECs and neutrophil nuclei were stained with 4′,6-diamidino-2-phenylindole (DAPI), and fixation was carried out with 4% formalin. Micrographs showing neutrophil adhesion were taken with an Eclipse 80i fluorescence microscope (Nikon, Champigny sur Marne, France) and analyzed using ImageJ.
Statistical analysis
Unless mentioned otherwise, quantitative variables were summarized as means and standard error of the mean. When the impact of different MP types incubated for different incubation periods were compared, we used 2-way analysis of variance, followed by Dunnett’s test (when there was a control AA MPs group) or Sidak’s test. Paired Student t tests were used to analyze the effects of preincubation of MPs with AV or lactadherin and ICAM-1 blocking antibody on ICAM-1–induced expression and neutrophil adhesion, respectively. Pearson’s correlation coefficient was used to assess the presence of correlations. The significance level was defined as P < .05. Analyses were conducted using GraphPad Prism 8 (GraphPad, La Jolla, CA).
Results
SS MPs increase endothelial ICAM-1 expression and subsequent neutrophil adhesion
To determine the effect of circulating SS, SC, or AA MPs on vascular endothelium, we incubated ECs with 3 doses of MPs (low, medium, and high); the last was representative of 7 MPs per cell (which is 4 times more than the medium dose and 20 times more than the low dose). After incubations of 2, 4, and 6 hours, we purified EC total messenger RNA (mRNA) to determine the levels of candidate mRNAs. No change was observed for HO-1 or PECAM-1 mRNAs. The inhibiting splicing isoform CD40 II31 was downregulated, and several mRNAs were overexpressed in SS vs AA MPs: ICAM-1 (Figure 1A), the signal transducing splicing isoform CD40 I, VCAM-1, and E-selectin (supplemental Figure 3). In contrast, compared with AA MPs, SC MPs only caused the overexpression of ICAM-1 (Figure 1A) and CD40 I (supplemental Figure 3). Moreover, compared with the low dose, the high dose caused an increase in ICAM-1 mRNA levels in the SC and SS groups, whereas a decrease was observed in the AA group (Figure 1A). There was no change at the protein level for VCAM-1 or E-selectin (data not shown) with SS or AA MPs. In contrast, endothelial ICAM-1 was overexpressed after 4 and 6 hours of incubation with SS MPs but not with SC or AA MPs (Figure 1B-C). The SS MP–mediated increase in ICAM-1 levels was inhibited by MP PS capping using AV or lactadherin (supplemental Figure 4). Importantly, the level of sICAM-1 observed in the plasma of SS patients was positively correlated with the in vitro ICAM-1–induced overexpression at 4 hours (r = +0.69 and P = .013 for MFI; r = +0.52 and P = .086 for positive cell percentage) and at 6 hours (r = +0.61 and P = .035 for MFI; r = +0.66 and P = .019 for positive cell percentage). Moreover, SS MPs also increased ICAM-1 expression in primary microvascular ECs to a greater extent compared with AA MPs (supplemental Figure 5).
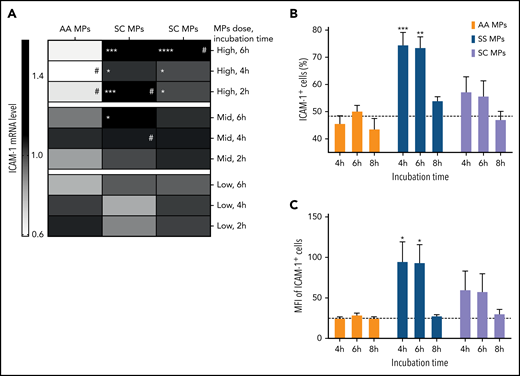
ICAM-1 mRNA and protein levels are increased by SS MPs. (A) Heat map indicating mean endothelial ICAM-1 mRNA level according to the dose of MPs, the incubation time, and the genotype of the subjects. Expression levels are normalized to the controls, without MPs. Flow cytometry analyses of a high dose of MPs and each considered genotype for each incubation time: the percentage of ICAM-1+ cells (B) and their MFI (C). Subjects per group: 9 ≤ n ≤ 12. The dotted lines represent the control level (without MPs) at 4 hours. #P < .05 vs corresponding low dose, *P < .05, **P < .01, ***P < .001, ****P < .0001 vs corresponding AA MP level.
ICAM-1 mRNA and protein levels are increased by SS MPs. (A) Heat map indicating mean endothelial ICAM-1 mRNA level according to the dose of MPs, the incubation time, and the genotype of the subjects. Expression levels are normalized to the controls, without MPs. Flow cytometry analyses of a high dose of MPs and each considered genotype for each incubation time: the percentage of ICAM-1+ cells (B) and their MFI (C). Subjects per group: 9 ≤ n ≤ 12. The dotted lines represent the control level (without MPs) at 4 hours. #P < .05 vs corresponding low dose, *P < .05, **P < .01, ***P < .001, ****P < .0001 vs corresponding AA MP level.
We subsequently incubated ECs with selected SS RBC-MPs or PLT-MPs for 4 hours and detected higher ICAM-1 protein levels with RBC-MPs (Figure 2), showing that RBC-MPs are more potent than PLT-MPs in triggering a proinflammatory phenotype in ECs. Because neutrophil adhesion to postcapillary venules is related, in part, to the overexpression of endothelial ICAM-1, we performed adhesion assays with neutrophils isolated from SCD blood samples. Fluorescently labeled SCD neutrophils were added to ECs that were preincubated with AA or SS MPs for 4 hours. Neutrophils adhered to ECs under both conditions, but SS MPs caused a 5.3-fold increase in the adhesion level compared with AA MPs (Figure 3).
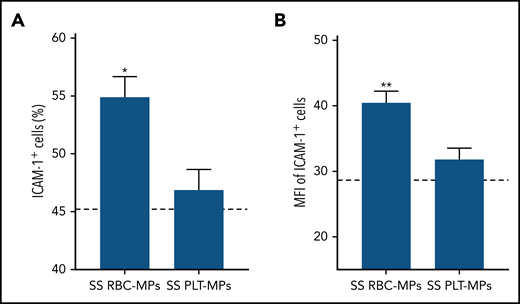
ICAM-1 overexpression is due to SS RBC-MPs rather than PLT-MPs. The percentage of ICAM-1+ cells (A) and their MFIs (B) were increased with SS RBC-MPs compared with the condition with PLT-MPs of the same SS patients (n = 9). The dashed lines represent the control level (without MPs) at 4 hours. *P < .05, **P < .01 vs SS PLT-MPs, paired Student t test.
ICAM-1 overexpression is due to SS RBC-MPs rather than PLT-MPs. The percentage of ICAM-1+ cells (A) and their MFIs (B) were increased with SS RBC-MPs compared with the condition with PLT-MPs of the same SS patients (n = 9). The dashed lines represent the control level (without MPs) at 4 hours. *P < .05, **P < .01 vs SS PLT-MPs, paired Student t test.
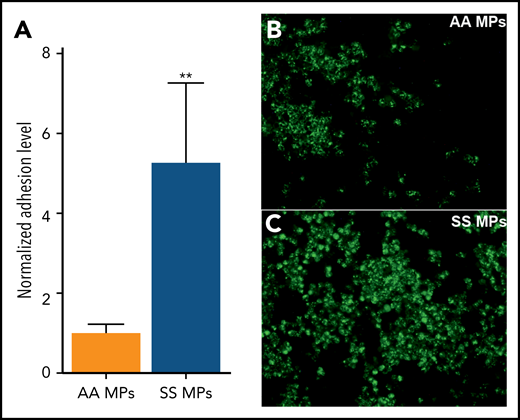
SS MPs increase neutrophil adhesion. (A) SCD neutrophil adhesion level was normalized to the one observed in the AA MP group. Representative photomicrographs taken during the same experiment, in a well in which cells were incubated with a high dose of AA MPs (B) or SS MPs (C) (n = 8 per group) (original magnification ×400). Neutrophils are fluorescently labeled (green). Nuclei of ECs and of neutrophils were labeled using DAPI (data not shown). **P < .01 vs AA MPs, Mann-Whitney U test.
SS MPs increase neutrophil adhesion. (A) SCD neutrophil adhesion level was normalized to the one observed in the AA MP group. Representative photomicrographs taken during the same experiment, in a well in which cells were incubated with a high dose of AA MPs (B) or SS MPs (C) (n = 8 per group) (original magnification ×400). Neutrophils are fluorescently labeled (green). Nuclei of ECs and of neutrophils were labeled using DAPI (data not shown). **P < .01 vs AA MPs, Mann-Whitney U test.
MPs preferentially binding to ECs exhibit high densities of exposed PS
Our group recently showed that the density of externalized PS at the surface of sickle MPs was diminished by HU treatment26 and increased during crisis.14 Therefore, we determined the PS density at the surface of MPs by flow cytometry. Total MPs, PLT-MPs, and RBC-MPs from the SS group had higher densities of externalized PS compared with the AA group, as estimated by the AV MFI of these MPs (supplemental Table 1). Next, we incubated the same quantities of SS MPs in parallel wells containing (EC+) or not containing (EC−) a monolayer of ECs. After 1 hour of incubation, MPs that did not bind to ECs, but remained in the supernatant, were analyzed using flow cytometry. Fewer MPs were detected in the supernatant of EC+ wells than in EC− wells for PLT-MPs and RBC-MPs (Figure 4A-B), indicating that a significant proportion of MPs bound to ECs. Interestingly, PS density of unbound MPs was lower in EC+ wells than in EC− wells (Figure 4C-D), whereas no difference in size (forward scatter values) was observed between the 2 conditions (Figure 4E-F), indicating preferential binding of MPs with higher levels of PS to ECs.
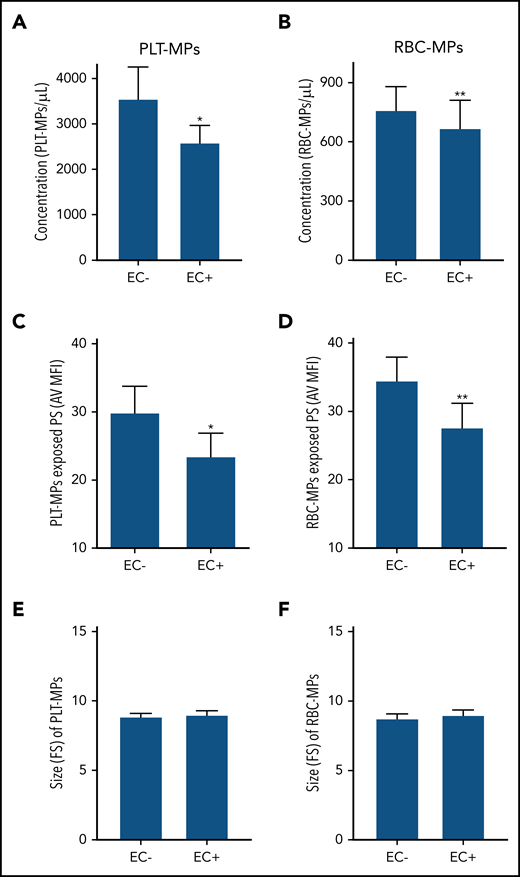
Involvement of externalized PS in SS MP binding to ECs. Concentration (A-B), PS exposure (C-D), and size (E-F) of SS PLT-MPs and SS RBC-MPs were determined after a 60-minute incubation with (EC+) or without (EC−) ECs (n = 17 patients per bar). *P < .05, **P < .01 vs EC−, Wilcoxon test. FS, forward scatter.
Involvement of externalized PS in SS MP binding to ECs. Concentration (A-B), PS exposure (C-D), and size (E-F) of SS PLT-MPs and SS RBC-MPs were determined after a 60-minute incubation with (EC+) or without (EC−) ECs (n = 17 patients per bar). *P < .05, **P < .01 vs EC−, Wilcoxon test. FS, forward scatter.
SS MP–mediated ICAM-1 induction is decreased by HU treatment and increased during crisis
We explored the impact of MPs on EC phenotype in 2 longitudinal cohorts. Consistent with our previous reports,14,26 externalized PS density was decreased by HU treatment (supplemental Table 2) and increased during crisis (supplemental Table 3). After 4 hours of incubation, endothelial ICAM-1 protein levels were lower with HU24 MPs than with HU0 MPs (Figure 5A-B). Opposite data were obtained during crisis, with higher ICAM-1 expression using crisis vs steady-state MPs (Figure 5C-D). We also detected a lower level of sICAM-1 in the supernatant when using HU24 MPs vs HU0 MPs but a higher level of sICAM-1 with crisis MPs vs steady-state MPs (supplemental Figure 6). Moreover, capping of PS of HU0 MPs and crisis MPs with AV decreased ICAM-1 (Figure 5) and sICAM-1 (supplemental Figure 6) levels compared with their corresponding uncapped MP conditions. We also observed the overexpression of ICAM-1 in primary human ECs using crisis MPs vs steady-state MPs (supplemental Figure 7).
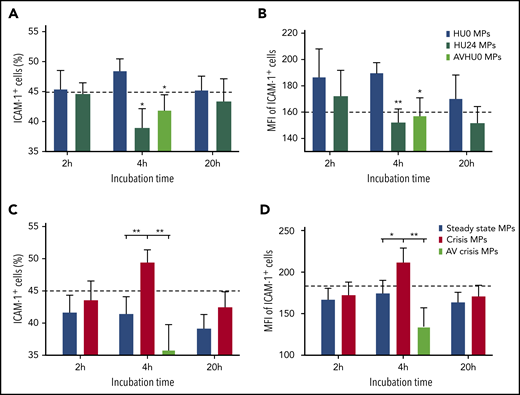
HU treatment or PS capping effects contrast with crisis MP–mediated increase in ICAM-1 level. At 4 hours, the percentage of ICAM-1+ cells (A) and their MFIs (B) were decreased with HU24 MPs or AVHU0 MPs (plasma MPs of untreated SS patients preincubated with AV to cap their exposed PS) compared with HU0 MPs (n = 15 for this cohort). *P < .05, **P < .01 vs corresponding HU0 MP level. For the other cohort of SS patients (n = 16), the percentage of ICAM-1+ cells (C) and their MFIs (D) were increased with crisis MPs vs steady-state MPs or AVcrisis MPs (plasma MPs of SS patients collected during crisis and preincubated with AV). Dotted lines represent control level (without MPs) at 4 hours. *P < .05, **P < .01.
HU treatment or PS capping effects contrast with crisis MP–mediated increase in ICAM-1 level. At 4 hours, the percentage of ICAM-1+ cells (A) and their MFIs (B) were decreased with HU24 MPs or AVHU0 MPs (plasma MPs of untreated SS patients preincubated with AV to cap their exposed PS) compared with HU0 MPs (n = 15 for this cohort). *P < .05, **P < .01 vs corresponding HU0 MP level. For the other cohort of SS patients (n = 16), the percentage of ICAM-1+ cells (C) and their MFIs (D) were increased with crisis MPs vs steady-state MPs or AVcrisis MPs (plasma MPs of SS patients collected during crisis and preincubated with AV). Dotted lines represent control level (without MPs) at 4 hours. *P < .05, **P < .01.
Neutrophil adhesion to ECs is decreased by HU24 MPs but increased by crisis MPs
To translate our findings regarding ICAM-1 expression into a functional approach, we performed sickle neutrophil adhesion assays. We observed a 2.8-fold decrease in the adhesion level with HU24 MPs vs HU0 MPs (Figure 6A-C). In contrast, we detected a 6.6-fold increase in adhesion levels with crisis MPs compared with steady-state MPs (Figure 6D-F). Finally, we showed that neutrophil adhesion relied on endothelial ICAM-1, because a blocking anti–ICAM-1 monoclonal antibody dramatically inhibited crisis MP–induced adhesion (Figure 6D-G), reaching a level that was even lower than in the steady-state group.
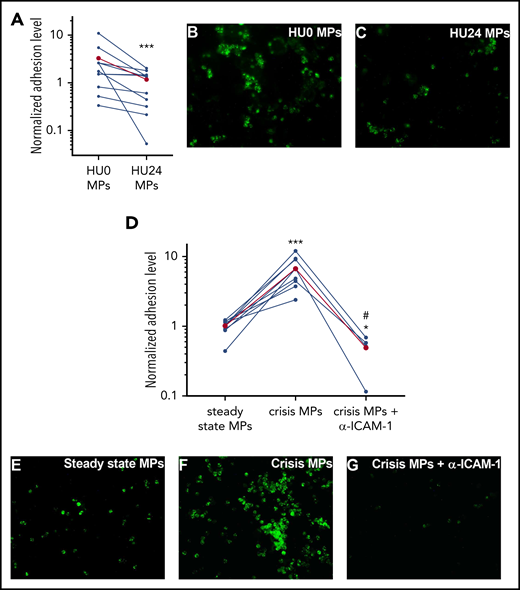
SCD neutrophil adhesion is decreased by HU24 MPs but potentiated by crisis MPs. After a 4-hour incubation of ECs with MPs, fluorescently labeled SCD neutrophils (green) had a significantly decreased adhesion level using HU24 MPs vs HU0 MPs (A), whereas crisis MPs increased the adhesion level compared with steady-state MPs from the same SS patients (n = 11 and 9 per cohort, respectively) (D). Combination of crisis MPs and blocking anti–ICAM-1 antibody (α–ICAM-1) led to a stark decrease in adhesion level compared with crisis MPs or steady-state MPs. Levels in the HU0 MPs or the steady-state MPs were used for normalization. The mean level for each group is shown in red. Representative photomicrographs of each condition are shown: HU0 MPs (B) or HU24 MPs (C) for the same SS patients or with steady-state MPs (E), crisis MPs (F), or crisis MPs from the same SS patients but in combination with a blocking anti–ICAM-1 antibody (G) (original magnification ×400). Nuclei of ECs and of neutrophils were labeled using DAPI (data not shown). #P < .05 vs crisis MPs, *P < .05, ***P < .001 vs HU0 MPs or steady-state MPs.
SCD neutrophil adhesion is decreased by HU24 MPs but potentiated by crisis MPs. After a 4-hour incubation of ECs with MPs, fluorescently labeled SCD neutrophils (green) had a significantly decreased adhesion level using HU24 MPs vs HU0 MPs (A), whereas crisis MPs increased the adhesion level compared with steady-state MPs from the same SS patients (n = 11 and 9 per cohort, respectively) (D). Combination of crisis MPs and blocking anti–ICAM-1 antibody (α–ICAM-1) led to a stark decrease in adhesion level compared with crisis MPs or steady-state MPs. Levels in the HU0 MPs or the steady-state MPs were used for normalization. The mean level for each group is shown in red. Representative photomicrographs of each condition are shown: HU0 MPs (B) or HU24 MPs (C) for the same SS patients or with steady-state MPs (E), crisis MPs (F), or crisis MPs from the same SS patients but in combination with a blocking anti–ICAM-1 antibody (G) (original magnification ×400). Nuclei of ECs and of neutrophils were labeled using DAPI (data not shown). #P < .05 vs crisis MPs, *P < .05, ***P < .001 vs HU0 MPs or steady-state MPs.
Discussion
In the present study, we showed that MPs isolated from sickle patients’ blood increased ICAM-1 endothelial expression at the RNA and protein levels and, consequently, the adhesion of neutrophils to these cells, a key event in the vaso-occlusive process.3,32 We also presented evidence that SS MPs proinflammatory properties varied according to the patients’ clinical condition, showing that MPs are biomarkers, as well as bioeffectors, in this disease.
We provided evidence that exposure of PS is involved in the proinflammatory properties of these extracellular vesicles. We observed that SS MPs with high PS exposure preferentially bound to ECs. This observation is in agreement with previous reports regarding the role of this phospholipid on the surfaces of RBCs33 and MPs.34,35 In addition, we showed that ICAM-1 overexpression was abolished after capping of SS MPs PS using AV or lactadherin. To gain insight into the impact of MPs from specific blood cells, we also selected the 2 most common MP subtypes. We found that SS RBC-MPs were more prone to induce ICAM-1 overexpression than were SS PLT-MPs, despite their relatively low concentration. However, ICAM-1 overexpression with total SS MPs may result from the synergy of RBC-MPs with other minor MP subtypes.25,34 Alternatively, the low concentration of RBC-MPs may also be compensated for by their higher exposure of PS compared with PLT-MPs,26,36 which may favor their binding to ECs and, thus, their biological impact on ECs. Our results with selected MPs contrast with previous reports showing that PLT-MPs mediated ICAM-1 overexpression.37,38 This discrepancy could be related to the use during the course of these 2 previous studies of higher doses of MPs, derived from platelets that were isolated from non-SCD subjects and produced using stimuli that may not recapitulate the in vivo conditions. The differences between ex vivo–generated MPs and circulating MPs is clearly illustrated in another study. Indeed, the incubation of ex vivo–generated RBC-MPs at only 50 MPs per microliter induced apoptosis in 40% of ECs, whereas a mean concentration of 637 RBC-MPs per microliter was reported in SCD plasma.39 Therefore, the relevance of our results relies on the use of circulating total MPs, which were incubated with ECs at a dose that was close to blood concentration, and on the positive correlation between ICAM-1 overexpression induced in vitro by SS MPs and plasma sICAM-1 levels observed in vivo in the same SS patients. In addition, a longitudinal study design allowed us to reduce the impact of interindividual variability in 2 of our 3 cohorts.
Strikingly, we observed increased neutrophil adhesion with SS MPs vs AA MPs, which supports the involvement of MPs in the proinflammatory context described in sickle patients.40 We also showed that crisis MPs increased ICAM-1 levels in a PS-dependent manner and dramatically increased SCD neutrophil adhesion. These MPs functional features may even have been underestimated because we used the same MP concentration in all cases, whereas MP concentration is known to be increased in SS plasma and even further during crisis.12,14 Adding an anti–ICAM-1 antibody inhibited neutrophil adhesion induced by crisis MPs. This is consistent with the prevention of vaso-occlusions observed in a murine model with the use of such an antibody.41 These findings support the potential use of an anti–ICAM-1 antibody in SCD patients. A P-selectin blocking antibody that was well tolerated during clinical trials42 prevents leukocyte rolling,43 which precedes ICAM-1–mediated firm neutrophil adhesion.7 Consequently, one might expect that an anti–ICAM-1 antibody might lead to even fewer adverse effects.
Aged neutrophils44 bearing 70% more Mac-1 protein (also known as αMβ2 or CD11b/CD18) are overrepresented in SS patients.45,46 This protein binds to endothelial ICAM-1 and allows the capture of sickle RBCs by neutrophils, which leads to VOC in mice.32,47-50 In line with these findings, our results collectively uncover a new mechanism of action of HU in SS patients, based on the reduction of MP PS exposure26 and the resulting inhibition of their proinflammatory properties (Figure 7). This study shows that there is a link between MP PS externalization and the induced proadhesive phenotype of ECs. We did not investigate the exact mechanisms responsible for intragroup or intergroup differences in MP PS density. However, erythroid oxidative stress could be involved, because it was shown to cause calcium entry into RBCs,51 which may promote membrane scrambling and PS externalization.52 Whatever the cause of MP PS density variations, it is noteworthy that outer PS might not have been sufficient to cause ICAM-1 overexpression, because SC MPs, which exposed even more PS than did SS MPs,36 did not trigger such an overexpression. Moreover, PS+ MP binding was not sufficient by itself to trigger ICAM-1 overexpression, because we showed that a larger number of PLT-MPs bound to ECs within 1 hour compared with RBC-MPs, without causing a comparable ICAM-1 overexpression. Nevertheless, PS was found to be involved in the overexpression caused by SS MPs, probably by permitting MPs to bind to ECs. Indeed, the use of AV to cap MP PS led to a reduction in ICAM-1 expression. This result suggests that AV, also called annexin A5 or placenta protein 4, might be a molecule to consider for SS patients. The side effects for such a potential treatment might be acceptable because this endogenous molecule has been studied extensively in human cell lines,53 animal models,54,55 and cancer patients,56,57 with no intolerable adverse effects reported.58 Alternatively, di-annexin,59 which has a longer lifespan in vivo and is used for patients with other diseases, could be considered as a potential therapeutic molecule in SCD.60-62

Impact of MPs on the proadhesive phenotype of ECs. PS exposure by plasma PLT-MPs and RBC-MPs, which may facilitate the binding of SS MPs to ECs, is increased during crisis but decreased after treatment with HU for 2 years. These changes in MP quality explain, at least in part, why MPs of HU-treated SS patients decreased ICAM-1 expression levels, whereas crisis MPs increased the expression of this adhesion protein. Neutrophil adhesion was consistently decreased using plasma MPs from HU-treated SS patients, whereas crisis MPs triggered increased adhesion. ICAM-1 overexpression and increased adhesion of neutrophils with crisis MPs were abolished by AV and ICAM-1 blocking, respectively. AA MPs cause a decrease in ICAM-1 at the mRNA and protein levels in ECs, as well as neutrophil adhesion, compared with MPs from untreated SS patients.
Impact of MPs on the proadhesive phenotype of ECs. PS exposure by plasma PLT-MPs and RBC-MPs, which may facilitate the binding of SS MPs to ECs, is increased during crisis but decreased after treatment with HU for 2 years. These changes in MP quality explain, at least in part, why MPs of HU-treated SS patients decreased ICAM-1 expression levels, whereas crisis MPs increased the expression of this adhesion protein. Neutrophil adhesion was consistently decreased using plasma MPs from HU-treated SS patients, whereas crisis MPs triggered increased adhesion. ICAM-1 overexpression and increased adhesion of neutrophils with crisis MPs were abolished by AV and ICAM-1 blocking, respectively. AA MPs cause a decrease in ICAM-1 at the mRNA and protein levels in ECs, as well as neutrophil adhesion, compared with MPs from untreated SS patients.
The present study had some limitations. We did not study the mechanism of interaction between MPs and ECs (ie, ligand-receptor interaction),63 MP fusion,34 or endocytosis.25 Consequently, we used the general term of “binding.” The static conditions used in our experiments did not allow us to correctly appreciate the impact of P-selectin, an endothelial protein playing a major role in the abnormal adherence of neutrophils in SCD.4 Furthermore, culturing ECs under low oxygen levels and with shear stress is known to modify some of their properties, as well as to increase their proinflammatory properties.64,65 In addition, we were only able to successfully detect MPs with a diameter >500 nm,66 which entails the underestimation of MP concentration. Several investigators consider vesicles of MP size with no exposed PS as MPs.67 The negligible number of such vesicles that we observed could be due to the low sensitivity of our flow cytometer. However, the fact that AV and lactadherin inhibited the proinflammatory properties of MPs suggests that these extracellular PS− vesicles are not involved in these properties.
The signaling pathway involved in the proinflammatory properties of sickle MPs remains to be understood. It has been shown that monocytic MPs contain interleukin-1β and induce endothelial ERK1/2 phosphorylation, leading to the production of various adhesive proteins, including ICAM-1.25 Interestingly, ERK1/2 is overexpressed and activated in SS RBCs but not in AA RBCs.68 Further studies are needed to decipher the signaling pathway activated by sickle MPs.
This study shows that circulating SS MPs, used at a relevant dose, triggered a PS-dependent ICAM-1 overexpression and, thereby, increased neutrophil adhesion, which is known to precede RBC adhesion69 and trapping in VOCs.40,70,71 Our data also support the notion that MPs have different effects on ECs, depending on their cell type of origin, as well as on the clinical status and the genotype of the individual. Altogether, our results show that circulating MPs are a new actor in the pathophysiology of SCD, which is targeted by HU treatment, and they point toward new possible therapeutics targeting the deleterious impact of MPs: AV and ICAM-1 blocking antibodies.
For original data, please contact the corresponding author.
The online version of this article contains a data supplement.
The publication costs of this article were defrayed in part by page charge payment. Therefore, and solely to indicate this fact, this article is hereby marked “advertisement” in accordance with 18 USC section 1734.
Acknowledgments
The authors thank Nathalie Vachiery and Valérie Pinarello for providing access to the Centre de Coopération Internationale en Recherche Agronomique pour le Développement (CIRAD) flow cytometer, Karubiotec for storing MPs, Olivier Gros for use of the fluorescence microscope at the Université des Antilles, and Eric Bailly for help with setting up the flow cytometry.
This work was supported by grants from the Conseil Regional de la Guadeloupe, the Conseil Regional de la Martinique, the French Ministry of Health, and Guadeloupe Espoir Drépanocytose (fellowship to Y.G.).
Authorship
Contribution: Y.G., M.G., A.C., and M.R. performed experiments; Y.G., S.F., K.-C.C., M.R., and R.H. purified MPs; Y.G. analyzed results and created the figures; M.R., P.C., M.-D.H.-D., C.L., Y.G., N.L., and M.E.-J. designed the research; and Y.G., M.R., W.E.N., and P.C. wrote the manuscript.
Conflict-of-interest disclosure: The authors declare no competing financial interests.
Correspondence: Marc Romana, UMR INSERM U1134, CHU de Pointe-à-Pitre, 97110 Pointe-à-Pitre, Guadeloupe, France; e-mail: marc.romana@inserm.fr.

This feature is available to Subscribers Only
Sign In or Create an Account Close Modal