

Key Points
ML NK cell differentiation and CAR engineering synergistically combine to enhance NK cell responses to resistant leukemia/lymphoma.
CAR-ML NK cells expand and control NK-resistant leukemia/lymphoma in vivo in xenograft models.

Abstract
Natural killer (NK) cells are a promising cellular immunotherapy for cancer. Cytokine-induced memory-like (ML) NK cells differentiate after activation with interleukin-12 (IL-12), IL-15, and IL-18, exhibit potent antitumor responses, and safely induce complete remissions in patients with leukemia. However, many cancers are not fully recognized via NK cell receptors. Chimeric antigen receptors (CARs) have been used to enhance tumor-specific recognition by effector lymphocytes. We hypothesized that ML differentiation and CAR engineering would result in complementary improvements in NK cell responses against NK-resistant cancers. To test this idea, peripheral blood ML NK cells were modified to express an anti-CD19 CAR (19-CAR-ML), which displayed significantly increased interferon γ production, degranulation, and specific killing against NK-resistant lymphoma lines and primary targets compared with nonspecific control CAR-ML NK cells or conventional CAR NK cells. The 19-CAR and ML responses were synergistic and CAR specific and required immunoreceptor tyrosine-based activation motif signaling. Furthermore, 19-CAR-ML NK cells generated from lymphoma patients exhibited improved responses against their autologous lymphomas. 19-CAR-ML NK cells controlled lymphoma burden in vivo and improved survival in human xenograft models. Thus, CAR engineering of ML NK cells enhanced responses against resistant cancers and warrants further investigation, with the potential to broaden ML NK cell recognition against a variety of NK cell–resistant tumors.
Introduction
Natural killer (NK) cells are cytotoxic innate lymphoid cells that are important for host responses against infection and mediate anticancer immune responses.1,2 NK cells directly kill diseased cells and communicate via cytokine and chemokine production.3 NK cells recognize potential target cells through the integration of signals by germ line DNA–encoded inhibitory, activating, and cytokine receptors2,4,5 and maintain self-tolerance through a licensing or education process that requires a self-recognizing inhibitory receptor.3 Advances over the past decade have revealed that NK cells mediate innate memory or memory-like (ML) responses after viral infection,6 certain hapten exposure,7 or cytokine activation specifically after a brief activation with interleukin-12 (IL-12), IL-15, and IL-18.8,9 Compared with conventional NK (cNK) cells, ML NK cells flexibly respond more potently to a variety of triggers, including cancer cells.9,10 In our first-in-human trial, donor ML NK cells safely induced clinical responses in 50% of poor-prognosis relapsed/refractory acute myeloid leukemia (AML) patients.10 However, not all patients respond to donor NK cells, and many types of cancer are not visible to NK cell receptors. Therefore, NK cell recognition of resistant blood cancers remains a barrier to the broad application of NK cell therapeutics.
Chimeric antigen receptor (CAR)–modified T cells targeting CD19 are effective for relapsed/refractory B-cell malignancies11-13 and are US Food and Drug Administration approved for relapsed/refractory diffuse large B-cell lymphoma and B-cell acute lymphoblastic leukemia.14-18 Along with its clinical success in inducing durable remissions, anti-CD19 CAR (19-CAR) T-cell therapy also exhibits significant adverse events associated with on-target/off-tumor effects, including severe inflammatory responses resulting from cytokine release syndrome (CRS) and immune cell–associated neurotoxicity (ICANS).19-21 Additional concerns with 19-CAR T cells include tumor-antigen escape22 and the long duration and high cost of cell therapy production. Therefore, there remains a clear need for additional innovations in engineered cellular therapy for cancer. Modification of NK cells with CARs are being investigated as an alternative to T cells.23,24 NK cells used in the allogeneic setting have an excellent adverse event profile and do not cause graft-versus-host disease (GVHD), CRS, or ICANS,25-27 including in a recent trial of cord blood (CB)–differentiated 19-CAR NK cells,28 which collectively suggests an improved safety profile for NK-based cellular therapy.
Here, we hypothesized that combining 2 strategies to enhance NK cell antileukemia/antilymphoma responses (ie, ML differentiation and CAR engineering) would synergistically improve recognition and response to NK-resistant targets, thereby overcoming barriers in the field. In this setting, ML NK cell function and persistence and CARs and endogenous NK cell–activating receptors work together to attack resistant blood cancers. We report proof-of-principal, preclinical experimental results demonstrating that human ML NK cells engineered to express 19-CARs (19-CAR-ML NK cells) have markedly enhanced responses to normally NK-resistant B-cell lymphoma malignancies in vitro and in vivo, thereby providing a novel strategy for blood cancer cellular therapy.
Materials and methods
Reagents, mice, and samples
The following recombinant human cytokines were used: recombinant human IL-12 (rhIL-12; Biolegend), rhIL-18 (InVivoGen), and rhIL-15 (Miltenyi). The Raji (American Type Culture Collection [ATCC] CCL-86), HL60 (AML), Kasumi-3 (Kas-3; ATCC CRL-2725), and MOLM-13 (AML-M5a) cell lines were used according to ATCC instructions. NSG (NOD-scid IL-2rγnull) mice (age 6-8 weeks) were obtained from The Jackson Laboratory, maintained under specific pathogen-free conditions, and used in accordance with our animal protocol approved by the Washington University Institutional Animal Care and Use Committee. Lymphoma patient samples provided were deidentified after collection on the Washington University Institutional Review Board–approved protocol (2011-08251).
NK cell purification and cell culture
Peripheral blood (PB) mononuclear cells were obtained from anonymous healthy platelet donors, and NK cells were purified using RosetteSep (StemCell Technologies; ≥95% CD56+CD3−). ML NK cells and control (conventional) NK cells were generated after in vitro stimulation with rhIL-12 (10 ng/mL), rhIL-18 (50 ng/mL), and rhIL-15 (50 ng/mL) or control conditions (rhIL-15; 1 ng/mL) as previously described.9,10
Construction of lentiviral vector and transduction of NK cells
The cassette encoding a single-chain variable fragment targeting CD19 (clone: fmc63),16 CD8α transmembrane, CD137, and CD3ζ was cloned into the MND lentiviral backbone29 to generate the CD19-CD8a-CD137-CD3ζ (19-CAR) vector. CD33-specific CARs (clone: my96; CD33-CD8a-CD137-CD3ζ) were similarly cloned.30 A third-generation packaging system pseudotyped with VSV-G in 293T cells produced lentivirus and was used to transduce NK cells. Lentivirus supernatants were added to 3 × 106 to 5 × 106 purified ML NK cells or cNK cells and spinfected at 30°C for 90 minutes on days 1 and 2. The cells were maintained in culture in complete media with the addition of rhIL-15 every 1 to 3 days.
Analysis of expression of CAR construct
To assess the surface expression of the CARs, cells were stained with an His-tagged human CD19 (Invitrogen), followed by staining with an APC anti-His tag antibody (J095G46; Biolegend). Cells were analyzed by flow cytometry for soluble CD19 molecules and green fluorescent protein (GFP; CAR+).
Functional and flow-based killing assay
CAR-ML NK cells were rested for 4 days after last spinfection to allow ML NK cell differentiation to occur. Cells were then harvested and stimulated with NK-resistant Raji targets, freshly thawed primary lymphoma targets, or CD33+ targets (effector/target ratio, 5:1, unless otherwise indicated) in the presence of 1 ng/mL of rhIL-15. Degranulation and interferon γ (IFN-γ) was assessed in 6-hour functional assays gated on GFP.9 For blocking experiments, Raji targets were preincubated with anti–human CD19 antibody (FCM63; 10 μg/mL) or isotype control (immunoglobulin G2; 10 μg/mL) for 30 minutes.
For cytotoxicity experiments, CAR-ML or control NK cells were coincubated with PKH67-labeled Raji or primary lymphoma cells for 4 hours, and 7-aminoactinomycin D staining was assessed by flow cytometry.31 The NK populations were not purified based on GFP expression, and effector/target ratios were calculated using total NK cells, not GFP+ NK cells.
Adoptive transfer of human CAR–transduced ML NK cells into NSG mice
NSG mice were irradiated with 250 cGy and inoculated IV with Raji cells (1 × 106) on day −3.32 On day 0, mice received IV injection of 3 × 106 to 5 × 106 total NK cells, containing 5% to 25% CAR+ 19-CAR-ML NK cells or 33-CAR-ML NK cells. To maintain NK cell survival in NSG mice that lacked homeostatic survival signals, rhIL-15 was injected intraperitoneally every 2 to 3 days. On day 18, the mice were euthanized, and spleen and blood were assessed for the presence of GFP+ NK cells. For bioluminescent imaging (BLI) and survival experiments, irradiated NSG mice were inoculated IV with luciferase-Raji cells (1 × 105) and 3 × 106 to 5 × 106 NK cells, containing 19-CAR-ML NK cells (mean ± standard error of the mean [SEM], 1.9 × 105 ± 0.4 × 105; range, 0.4 × 105 to 3.9 × 105) or 33-CAR-ML NK cells (mean ± SEM, 1.9 × 105 ± 0.4 × 105; range, 0.7 × 105 to 4.0 × 105). Mice were evaluated by BLI to assess tumor burden and followed for survival (supplemental Methods, available on the Blood Web site).
Flow cytometry
Flow cytometry was performed on a Gallios (Beckman Coulter) and analyzed using FlowJo (Tree Star) software as described.9,10 Cell sorting was performed using a FACSAria II cell sorter (BD Biosciences).
Mass cytometry and data analysis
Donor NK cells were assessed by mass cytometry (supplemental Table 1). All samples were processed and analyzed using Cytobak as previously described.10 Flow-sorted GFP− ML cells and GFP+ CAR-transduced ML NK cells were analyzed using visualization of t-distributed stochastic neighbor embedding (viSNE).
Statistical analysis
Between-group differences (mean ± SEM) were compared using the paired Student t test, 2-sample Student t test, or analysis of variance as appropriate. Analyses were performed using Prism v8. P values were 2 sided, and P < .05 was considered significant. The synergic effect of ML differentiation and CAR expression in the overall response of NK cell was calculated by the combination index (CI)33 (supplemental Methods).
Results
CAR lentiviral vectors effectively transduce human PB ML NK cells
Although preclinical studies have demonstrated that CB cells28 and induced pluripotent stem cells (iPSCs)23 may be transduced with CAR lentiviral or retroviral vectors and differentiated into NK cells over weeks, the feasibility of engineering human PB ML NK cells with CAR constructs and the subsequent antitumor potency of these cells have not yet been established. To address this, we used single-chain variable fragments targeting CD19 or CD33 to generate a second-generation CAR with CD137 and CD3ζ intracellular signaling domains (Figure 1A). Both CD13734,35 and CD3ζ36,37 were chosen because they are used by endogenous NK cells for signaling and activation. We also generated a truncated 19-CAR molecule (19-CARtrunc) that lacked ITAM signaling domains. CAR expression by NK cells was measured by expression of P2A-GFP in these preclinical CAR constructs. Purified PB NK cells were activated with IL-12, IL-15, and IL-18 to generate ML NK cells or IL-15 to maintain survival, resulting in control cNK cells (Figure 1B),9 transduced with lentivirus, and cultured for a total of 5 to 7 days to allow for ML differentiation and CAR expression. There was no significant difference in transduction efficiency between cNK cells or ML NK cells with this approach, but a modest trend toward higher transduction efficiency with ML NK cells was seen (Figure 1C). Additionally, 19-CAR-ML NK cells stained with a soluble CD19 molecule showed concordant surface expression of 19-CARs with GFP expression (Figure 1D).
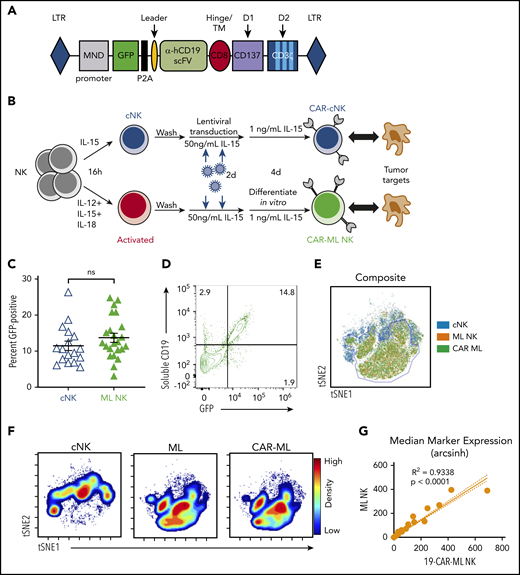
Human ML NK cells are effectively transduced with CAR lentiviral vectors. (A) Schematic representation of the lentiviral cassette encoding the αCD19 CAR. P2A indicates the ribosomal P2A skip site. Transmembrane (TM)/hinge: CD8α; costimulatory domain (D1): CD137; and stimulatory domain (D2): CD3ζ. ITAMs indicated in light blue. (B) Schema of in vitro experiments. Purified NK cells were activated with IL-12, IL-15, and IL-18 or were control treated for 16 hours, washed, and transduced with CAR lentivirus for 2 days. After differentiating for 1 week, NK cell phenotype and functionality were assessed. (C) Summary of data of viral transduction efficiency between low-dose cNK cells (blue open triangle) and ML NK cells (green triangle; n = 17-23 donors from 15 independent experiments; each symbol represents 1 donor; line indicates mean ± SEM). Data were compared using paired Student t test. (D) Representative bivariate flow plots showing expression of GFP and sCD19 staining on 19-CAR-ML NK cells. (E) Representative overlaid viSNE plot of cNK (blue), ML (orange), and CAR-ML NK cells (green). (F) Representative viSNE density plot of cNK, ML NK, and CAR-ML NK cells. (G) Linear regression model of phenotypic markers of 19-CAR-ML NK cells (x-axis) and ML NK cells (y-axis). hCD19, human CD19; LTR, long terminal repeat; ns, no significance; scFv, single-chain variable fragment.
Human ML NK cells are effectively transduced with CAR lentiviral vectors. (A) Schematic representation of the lentiviral cassette encoding the αCD19 CAR. P2A indicates the ribosomal P2A skip site. Transmembrane (TM)/hinge: CD8α; costimulatory domain (D1): CD137; and stimulatory domain (D2): CD3ζ. ITAMs indicated in light blue. (B) Schema of in vitro experiments. Purified NK cells were activated with IL-12, IL-15, and IL-18 or were control treated for 16 hours, washed, and transduced with CAR lentivirus for 2 days. After differentiating for 1 week, NK cell phenotype and functionality were assessed. (C) Summary of data of viral transduction efficiency between low-dose cNK cells (blue open triangle) and ML NK cells (green triangle; n = 17-23 donors from 15 independent experiments; each symbol represents 1 donor; line indicates mean ± SEM). Data were compared using paired Student t test. (D) Representative bivariate flow plots showing expression of GFP and sCD19 staining on 19-CAR-ML NK cells. (E) Representative overlaid viSNE plot of cNK (blue), ML (orange), and CAR-ML NK cells (green). (F) Representative viSNE density plot of cNK, ML NK, and CAR-ML NK cells. (G) Linear regression model of phenotypic markers of 19-CAR-ML NK cells (x-axis) and ML NK cells (y-axis). hCD19, human CD19; LTR, long terminal repeat; ns, no significance; scFv, single-chain variable fragment.
Mass cytometry reveals unbiased CAR lentiviral transduction of ML NK cells
Because PB human NK cells were incompletely transduced by this lentivirus approach, we questioned whether a subset of NK cells was preferentially transduced, which could have biased downstream analyses and functional assays. To test this, an established mass cytometry panel9 (supplemental Table 1) that included NK cell lineage and maturation markers, inhibitory and activating receptors, and function-associated molecules was used. Sorted GFP− (CAR−) and GFP+ (CAR+) 19-CAR-ML NK cells (>96% purity) were generated and assessed by mass cytometry using unsupervised t-distributed SNE–based clustering. The location of cNK cells on the viSNE map was clearly distinct from both ML NK cells and CAR-ML NK cells (Figure 1E-F). In contrast, ML and CAR-ML NK cells overlapped within the viSNE plot, with very high concordance of the expression of the assessed markers (Figure 1E-G). There was also no significant bias in activating receptors, inhibitory receptors, or maturation stage subsets (Figure 1G; supplemental Figure 1), indicating that ML NK cells transduced with CARs share the repertoire and subset diversity of nontransduced ML NK cells.
CAR-ML NK cells exhibit enhanced functional responses compared with CAR cNK cells
To understand how ML differentiation and CAR expression contributed to antilymphoma responses, 19-CAR-ML NK cells were directly compared with 19-CAR cNK cells. 19-CAR-ML NK and 19-CAR cNK cells from the same donors were evaluated against Raji cell targets (Figure 2A-B; supplemental Figure 2). 19-CAR cNK cells demonstrated significantly increased IFN-γ production and degranulation against Raji targets compared with cNK cells (19-CAR−; Figure 2B-D). Regarding the ML NK cells (19-CAR−), we observed a modest but significant increase in IFN-γ response to Raji targets compared with cNK cells (19-CAR−). 19-CAR-ML NK cells demonstrated superior IFN-γ and degranulation compared with all other conditions (Figure 2B-D). Because both ML differentiation and CAR expression individually improved functional responses against Raji targets, we evaluated for synergistic interactions between those 2 attributes9 using the CI.33,38 CI values were derived from mean percentage of IFN-γ− and CD107a+ cells and used to determine whether the CI values were significantly different from the CI value of 1 (defined as no synergy; “Materials and methods”). The results showed a significant beneficial synergism for both IFN-γ (CI, 0.587; P = .040) and CD107a (CI, 0.503; P = .024), indicating synergism between CAR and ML differentiation for enhanced NK cell function (Figure 2E). 19-CAR-ML NK cells were next compared with 19-CAR cNK cells for their ability to kill Raji targets in a flow-based killing assay. 19-CAR-ML NK cells exhibited increased killing of Raji targets compared with donor-matched 19-CAR cNK cells (Figure 2F). Thus, both ML differentiation and CAR engineering combine to maximize the antilymphoma effector functions and cytotoxicity against CD19+ targets by 19-CAR-ML NK cells.
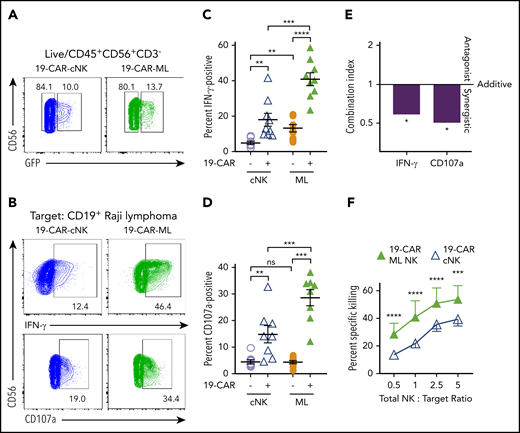
CAR-ML NK cells exhibit enhanced functional responses compared with CAR cNK cells. cNK cells or ML NK cells were transduced with 19-CAR lentivirus and effector functions against CD19+ Raji targets were assessed 7 days later. (A) Representative GFP gating strategy from cNK (blue) and ML NK cells (green) transduced with 19-CAR lentivirus. (B) Representative flow plots of GFP+ cNK or ML NK cells stimulated with Raji targets (total NK/T, 5:1) depicting IFN-γ (top) and degranulation (CD107a; bottom). (C) Summary IFN-γ production from panel B. (D) Summary degranulation from panel B (n = 8-9; 5 independent experiments). (E) Combination index values of IFN-γ and CD107a from panels C and D. (F) Summary data show mean ± SEM percentage of specific killing by 19-CAR cNK and 19-CAR-ML NK cells at the indicated total NK effector/target ratios (n = 4; 2 independent experiments). cNK cells, open lavender circle; ML NK cells, orange circle; 19-CAR cNK cells, blue open triangle, 19-CAR ML NK cells, green triangle. Data were compared using 2-way analysis of variance with Bonferroni’s multiple comparisons test (C-E) or linear mixed model with Bonferroni’s multiple comparisons test (F). *P < .05, **P < .01, ***P < .001, ****P < .0001. ns, no significance.
CAR-ML NK cells exhibit enhanced functional responses compared with CAR cNK cells. cNK cells or ML NK cells were transduced with 19-CAR lentivirus and effector functions against CD19+ Raji targets were assessed 7 days later. (A) Representative GFP gating strategy from cNK (blue) and ML NK cells (green) transduced with 19-CAR lentivirus. (B) Representative flow plots of GFP+ cNK or ML NK cells stimulated with Raji targets (total NK/T, 5:1) depicting IFN-γ (top) and degranulation (CD107a; bottom). (C) Summary IFN-γ production from panel B. (D) Summary degranulation from panel B (n = 8-9; 5 independent experiments). (E) Combination index values of IFN-γ and CD107a from panels C and D. (F) Summary data show mean ± SEM percentage of specific killing by 19-CAR cNK and 19-CAR-ML NK cells at the indicated total NK effector/target ratios (n = 4; 2 independent experiments). cNK cells, open lavender circle; ML NK cells, orange circle; 19-CAR cNK cells, blue open triangle, 19-CAR ML NK cells, green triangle. Data were compared using 2-way analysis of variance with Bonferroni’s multiple comparisons test (C-E) or linear mixed model with Bonferroni’s multiple comparisons test (F). *P < .05, **P < .01, ***P < .001, ****P < .0001. ns, no significance.
19-CAR-ML NK cells exhibit CD19-specific responses to CD19+ targets requiring CAR signaling
Next, the specificity of 19-CAR-ML NK cells for the CD19 antigen was investigated using CD19+ Raji targets vs CD19− Kas-3 targets. 19-CAR-ML NK cells exhibited a fourfold increase in IFN-γ production and sixfold increase in degranulation against Raji targets compared with ML NK cells, but minimal changes against Kas-3 targets (Figure 3A-B). To confirm antigen specificity, ML NK cells transduced with a control 33-CAR-ML lentivirus did not have enhanced responses to Raji targets (CD19+CD33−) compared with nontransduced ML NK cells (Figure 3C-D), although they displayed enhanced functional responses to a variety of CD33+ targets (supplemental Figure 3). Additionally, monoclonal antibody blockade of CD19 resulted in abrogation of the 19-CAR-ML NK cell response to control levels (supplemental Figure 3E-F). Furthermore, Raji killing was increased in the 19-CAR-ML NK cell condition compared with 33-CAR-ML NK cell controls, even at low CAR+ effector cell/target ratios of <1:1 (range, 0.05:1 to 0.75:1; Figure 3E; supplemental Figure 4). To determine if the CAR intracellular signaling component was required for enhanced antitumor responses, stimulation of a 19-CARtrunc-ML with Raji targets abolished the increase in IFN-γ production and degranulation observed in 19-CAR-ML NK cells (Figure 3F-G). Together, these data demonstrate that 19-CAR-ML NK cells’ enhanced responses were CAR antigen specific and required intracellular CAR ITAM signaling.
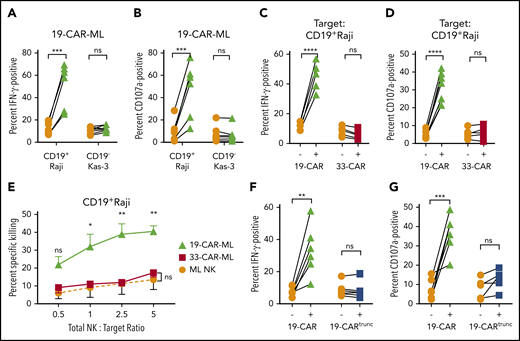
19-CAR-ML NK cells display enhanced antigen (CD19)–specific responses against tumor cell lines. (A-B) 19-CAR-ML NK cells were incubated with CD19+ Raji or CD19− Kas-3 target cells for 6 hours at a 5:1 total NK/T ratio. Summary data show percentage of IFN-γ (A) and CD107a+ positive cells (B) (n = 7; 3 independent experiments). (C-D) ML NK, 19-CAR-ML NK, and 33-CAR-ML NK cells were incubated with Raji for 6 hours at a 5:1 total NK/T ratio. Summary data show IFN-γ production (C) and CD107a expression (D) (n = 5-6; 4 independent experiments). (E) 19-CAR-ML NK, 33-CAR-ML NK, and ML NK cells were assessed for cytotoxicity against Raji cells in a 4-hour flow-based killing assay. Summary data show mean ± SEM percentage of specific killing by 19-CAR-ML NK cells (green triangle), 33-CAR-ML NK cells (red square), and ML NK cells (orange circle; n = 4; 3 independent experiments). (F) 19-CARtrunc-ML NK cells were incubated with Raji for 6 hours at 5:1 total NK/T ratio. Summary data show percentage of (F) IFN-γ (F) and CD107a (G) (n = 5-6; 3 independent experiments). Data were compared using paired Student t test (A-D,F-G) or linear mixed model with Bonferroni’s multiple comparisons test (E). *P < .05, **P < .01, ***P < .001, ****P < .0001. ns, no significance.
19-CAR-ML NK cells display enhanced antigen (CD19)–specific responses against tumor cell lines. (A-B) 19-CAR-ML NK cells were incubated with CD19+ Raji or CD19− Kas-3 target cells for 6 hours at a 5:1 total NK/T ratio. Summary data show percentage of IFN-γ (A) and CD107a+ positive cells (B) (n = 7; 3 independent experiments). (C-D) ML NK, 19-CAR-ML NK, and 33-CAR-ML NK cells were incubated with Raji for 6 hours at a 5:1 total NK/T ratio. Summary data show IFN-γ production (C) and CD107a expression (D) (n = 5-6; 4 independent experiments). (E) 19-CAR-ML NK, 33-CAR-ML NK, and ML NK cells were assessed for cytotoxicity against Raji cells in a 4-hour flow-based killing assay. Summary data show mean ± SEM percentage of specific killing by 19-CAR-ML NK cells (green triangle), 33-CAR-ML NK cells (red square), and ML NK cells (orange circle; n = 4; 3 independent experiments). (F) 19-CARtrunc-ML NK cells were incubated with Raji for 6 hours at 5:1 total NK/T ratio. Summary data show percentage of (F) IFN-γ (F) and CD107a (G) (n = 5-6; 3 independent experiments). Data were compared using paired Student t test (A-D,F-G) or linear mixed model with Bonferroni’s multiple comparisons test (E). *P < .05, **P < .01, ***P < .001, ****P < .0001. ns, no significance.
19-CAR-ML NK cells have enhanced responses against patient primary lymphoma cells
Next, the antitumor activity of 19-CAR-ML NK cells against CD19+ primary lymphoma in vitro was investigated. First, we examined the functional responses by 19-CAR-ML NK cells and 19-CAR cNK cells when incubated with primary lymphoma cells (supplemental Table 2). Significantly increased IFN-γ production and degranulation against primary patient lymphoma cells by 19-CAR-ML NK cells were observed, compared with 19-CAR cNK cells (Figure 4A). Consistent with responses to Raji cells (Figure 3), the enhanced 19-CAR-ML NK cell responses against primary lymphoma targets were CAR antigen specific and were not observed with 33-CAR-ML NK cells (Figure 4B). Cytotoxicity against primary lymphoma targets by 19-CAR-ML NK cells and 33-CAR-ML NK cells was tested, and significantly improved killing by 19-CAR-ML NK cells compared with 33-CAR-ML NK cells was observed (Figure 4C). Differences in analyzing gated CAR+ NK cells vs bulk NK cells likely account for differences in the magnitude of degranulation vs cytotoxicity (“Materials and methods”). Thus, 19-CAR-ML NK cells have enhanced CAR antigen–specific antitumor responses against primary lymphoma targets.
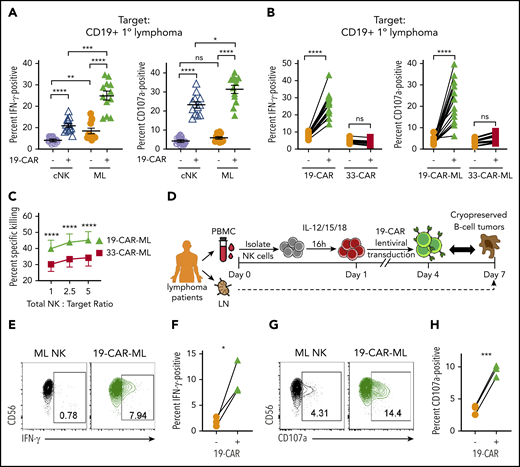
19-CAR-ML NK cells display enhanced responses to primary lymphoma targets in vitro. (A) 19-CAR cNK and 19-CAR-ML NK cells were incubated with CD19+ B-cell lymphoma tumor cells for 6 hours at 5:1 total NK/T ratio. Summary data shown as scatter dot plot of percentage of IFN-γ and CD107a+ cells with line at mean ± SEM (n = 12; 3 independent experiments). (B) 19-CAR-ML and 33-CAR-ML NK cells were incubated with CD19+ B-cell lymphoma tumors for 6 hours at a 5:1 total NK/T ratio. Summary data show percentage of IFN-γ and CD107a+ cells (n = 8-13; 4-6 independent experiments). (C) Summary primary lymphoma killing assay showing mean ± SEM percentage of specific killing by 19-CAR-ML and 33-CAR-ML NK cells at the indicated effector/target ratios (n = 3-4; 2-3 independent experiments). (D) Experimental schema. Briefly, PB samples were obtained from patients with B-cell lymphoma at day 0. NK cells were isolated, activated with IL-12, IL-15, and IL-18, and transduced with 19-CAR lentivirus. On day 7, 19-CAR-ML NK cells were incubated with autologous B-cell lymphoma tumor for 6 hours at a 5:1 Total NK/T ratio. (E,G) Representative bivariate flow cytometry plots show percentage of NK cells positive for IFN-γ (E) and CD107a (G) in response to autologous tumor targets. (F,H) Summary data show percentage of IFN-γ (F) and CD107a+ cells (H) of autologous 19-CAR-ML NK cells and control ML NK cells (n = 3 patients; 2 independent experiments). Data were compared using 2-way analysis of variance with Bonferroni’s multiple comparisons test (A,C) or paired Student t test (B,F,H). *P < .05, **P < .01, ***P < .001, ****P < .0001. ns, no significance.
19-CAR-ML NK cells display enhanced responses to primary lymphoma targets in vitro. (A) 19-CAR cNK and 19-CAR-ML NK cells were incubated with CD19+ B-cell lymphoma tumor cells for 6 hours at 5:1 total NK/T ratio. Summary data shown as scatter dot plot of percentage of IFN-γ and CD107a+ cells with line at mean ± SEM (n = 12; 3 independent experiments). (B) 19-CAR-ML and 33-CAR-ML NK cells were incubated with CD19+ B-cell lymphoma tumors for 6 hours at a 5:1 total NK/T ratio. Summary data show percentage of IFN-γ and CD107a+ cells (n = 8-13; 4-6 independent experiments). (C) Summary primary lymphoma killing assay showing mean ± SEM percentage of specific killing by 19-CAR-ML and 33-CAR-ML NK cells at the indicated effector/target ratios (n = 3-4; 2-3 independent experiments). (D) Experimental schema. Briefly, PB samples were obtained from patients with B-cell lymphoma at day 0. NK cells were isolated, activated with IL-12, IL-15, and IL-18, and transduced with 19-CAR lentivirus. On day 7, 19-CAR-ML NK cells were incubated with autologous B-cell lymphoma tumor for 6 hours at a 5:1 Total NK/T ratio. (E,G) Representative bivariate flow cytometry plots show percentage of NK cells positive for IFN-γ (E) and CD107a (G) in response to autologous tumor targets. (F,H) Summary data show percentage of IFN-γ (F) and CD107a+ cells (H) of autologous 19-CAR-ML NK cells and control ML NK cells (n = 3 patients; 2 independent experiments). Data were compared using 2-way analysis of variance with Bonferroni’s multiple comparisons test (A,C) or paired Student t test (B,F,H). *P < .05, **P < .01, ***P < .001, ****P < .0001. ns, no significance.
Lymphoma patient 19-CAR-ML NK cells respond to autologous lymphoma cells
We next investigated whether lymphoma patient NK cells could be engineered to respond against their own lymphoma cells. 19-CAR-ML NK cells from lymphoma patients were generated and tested for functional responses (Figure 4D). Although patient ML NK cells responded weakly to autologous lymphomas targets (Figure 4E-H), patient 19-CAR-ML NK cells produced significantly increased IFN-γ and degranulation. Thus, the addition of targeted CAR molecules to patient ML NK cells results in significant improvement in autologous antitumor NK responses.
19-CAR-ML NK cells have CAR antigen–specific expansion in vivo
Previous studies have established that after IL-12, IL-15, and IL-18 activation, ML NK cells differentiate within NSG mice in vivo, and similar to cNK cells, their survival in mouse xenograft models requires rhIL-15 or rhIL-2.8,9 Using this experimental system, Raji cells were engrafted in NSG mice, and 19-CAR-ML and 33-CAR-ML NK cells were adoptively transferred into Raji-bearing mice (Figure 5A). On day 18 after NK cell transfer, 19-CAR-ML NK cells accounted for 40% to 90% of the NK cell population in vivo in blood in the presence of Raji, a significant increase compared with 33-CAR-ML–recipient mice (Figure 5B-D). This is substantially higher than the average transduction efficiency after in vitro differentiation without CD19 targets (Figure 1C), suggesting expansion of CAR+ ML NK cells in vivo. These data suggest that CAR-mediated CD137/CD3ζ signals contribute to NK cell survival and/or expansion in the presence of CD19-expressing targets in vivo. In this experiment, and in other in vivo experiments in this study, we focused our comparisons on CD33-CAR-ML (control) vs CD19-CAR-ML (experimental) NK cells as the most relevant to establish the translational potential of CD19-CAR-ML NK cells. This was based in part on the prior work demonstrating that ML NK cells exhibited superior responses to myeloid leukemia targets compared with cNK cells.8,9 Furthermore, in vivo CAR-ML NK cells expand in vivo after brief transduction and become 40% to 90% CAR+, suggesting that relatively modest lentivirus transduction yields adequate CAR-ML NK cells for in vivo activity.
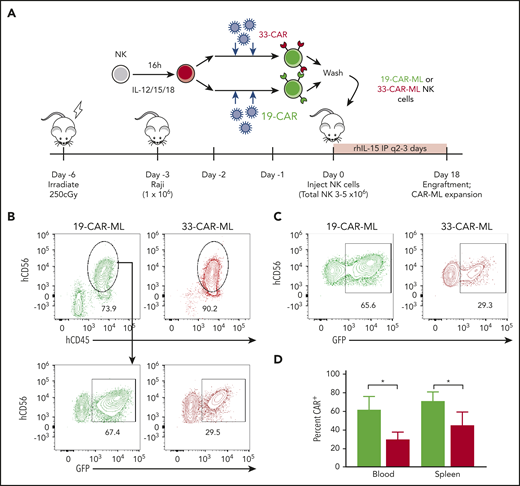
19-CAR-ML NK cells have antigen-specific persistence in vivo. (A) Schema of in vivo studies. Briefly, NSG mice received 1 × 106 Raji cells IV on day −3 followed by 3 × 106 to 5 × 106 19-CAR-ML NK or 33-CAR-ML NK cells IV on day 0. Mice were euthanized on day 18, and human NK (hNK; mCD45−hCD45+CD56+) cells in the spleen and blood were assessed. (B) Representative bivariate plots at day 18 after transfer showing 19-CAR-ML (green) or 33-CAR-ML (red) NK cells in the blood. (C) Representative bivariate plots at day 18 after transfer showing 19-CAR-ML (green) or 33-CAR-ML (red) NK cells in the spleen, gated as in panel B. (D) Summary of data demonstrating the percentages of GFP+ NK cells in each organ. Data are from 2 experiment with n = 5 mice per group represented as mean ± SEM, compared using paired Student t test. *P < .05.
19-CAR-ML NK cells have antigen-specific persistence in vivo. (A) Schema of in vivo studies. Briefly, NSG mice received 1 × 106 Raji cells IV on day −3 followed by 3 × 106 to 5 × 106 19-CAR-ML NK or 33-CAR-ML NK cells IV on day 0. Mice were euthanized on day 18, and human NK (hNK; mCD45−hCD45+CD56+) cells in the spleen and blood were assessed. (B) Representative bivariate plots at day 18 after transfer showing 19-CAR-ML (green) or 33-CAR-ML (red) NK cells in the blood. (C) Representative bivariate plots at day 18 after transfer showing 19-CAR-ML (green) or 33-CAR-ML (red) NK cells in the spleen, gated as in panel B. (D) Summary of data demonstrating the percentages of GFP+ NK cells in each organ. Data are from 2 experiment with n = 5 mice per group represented as mean ± SEM, compared using paired Student t test. *P < .05.
19-CAR-ML NK cells control lymphoma targets in vivo
Next, we examined the impact of CAR expression on ML NK cell effector functions in vivo. NSG mice were infused IV with luciferase-expressing Raji cells and a single injection of 1.9 × 105 19-CAR–transduced ML NK cells or 33-CAR–transduced ML NK cells (3 × 106 to 5 × 106 total NK cells; Figure 6A). After adoptive transfer, low-dose rhIL-15 was administered to support in vivo NK survival. Tumor burden was monitored by BLI.32 Compared with both untreated tumor-bearing mice and 33-CAR-ML NK cell–treated mice, treatment with 19-CAR-ML NK cells resulted in a significant decrease in tumor burden (Figure 6B-D). The enhanced antitumor control after a single injection of 19-CAR-ML NK cells also resulted in improved survival of treated mice compared with 33-CAR-ML NK cells (Figure 6E). These data demonstrated that a 19-CAR-ML NK cell product containing 10% to 25% CAR+ NK cells provides enhanced tumor control in vivo.

19-CAR-ML NK cells effectively control lymphoma targets in vivo in NSG mice. (A) Schema of in vivo studies using luciferase (luc)-expressing Raji. Irradiated mice received 1 × 105 Raji-luc cells IV followed by 1.9 × 105 33-CAR-ML or 19-CAR-ML NK cells (total, 3 × 106 to 5 × 106 IV on day 0). Lymphoma burden was determined by every-other-day BLI. (B) Representative BLI images from mice at indicated time points. (C) Summary BLI data of tumor burden of each group on day 24. Day 24 data are presented, because this was the longest time point that included all the mice in the untreated group before mortality from lymphoma. Data shown as bar graph with mean ± SEM (n = 7-11 total mice from 2 independent experiments). (D) Summary of BLI data of the tumor burden of each group monitored for 28 days after cell infusion. Mean ± standard deviation shown (n = 10-11; 2 independent experiments) (E) Kaplan-Meier survival curve of mice receiving tumor only (blue line), 33-CAR-ML NK cells (red line), or 19-CAR-ML NK cells (green line). Data were compared with a linear mixed model followed by Student t test for 19-CAR-ML vs 33-CAR-ML NK cells (D) and log-rank test (E). *P < .05, **P < .01, ***P < .001, ****P < .0001.
19-CAR-ML NK cells effectively control lymphoma targets in vivo in NSG mice. (A) Schema of in vivo studies using luciferase (luc)-expressing Raji. Irradiated mice received 1 × 105 Raji-luc cells IV followed by 1.9 × 105 33-CAR-ML or 19-CAR-ML NK cells (total, 3 × 106 to 5 × 106 IV on day 0). Lymphoma burden was determined by every-other-day BLI. (B) Representative BLI images from mice at indicated time points. (C) Summary BLI data of tumor burden of each group on day 24. Day 24 data are presented, because this was the longest time point that included all the mice in the untreated group before mortality from lymphoma. Data shown as bar graph with mean ± SEM (n = 7-11 total mice from 2 independent experiments). (D) Summary of BLI data of the tumor burden of each group monitored for 28 days after cell infusion. Mean ± standard deviation shown (n = 10-11; 2 independent experiments) (E) Kaplan-Meier survival curve of mice receiving tumor only (blue line), 33-CAR-ML NK cells (red line), or 19-CAR-ML NK cells (green line). Data were compared with a linear mixed model followed by Student t test for 19-CAR-ML vs 33-CAR-ML NK cells (D) and log-rank test (E). *P < .05, **P < .01, ***P < .001, ****P < .0001.
Discussion
Here, we developed a process to generate ML NK cells with stable CAR expression from PB NK cell in 5 to 7 days. These CAR-ML NK cells induce potent responses against NK-resistant cancer targets with a notable synergistic combination of ML differentiation and CAR-mediated targeting. We established that the enhanced CAR-ML NK cell functional responses (cytotoxicity, degranulation, and cytokine production) were CAR antigen specific and required CAR signaling. These enhanced effector responses were observed against NK-resistant lymphoma lines, as well as against primary lymphoma cell targets. Relevant to future translation, 19-CAR-ML NK cells generated from lymphoma patient NK cells had improved antitumor activity against autologous lymphoma targets. Moreover, 19-CAR-ML NK cells were able to effectively expand and persist, control tumor growth, and prolong survival of tumor-bearing mice in an NSG xenograft model, used previously for NK cell32 and CB-differentiated CAR NK cell assessments.24 Thus, the development of CAR-ML NK cells represents a novel approach to CAR-engineered NK cellular immunotherapy and warrants further preclinical and clinical investigation.
NK cells represent a nascent approach for cancer immunotherapy and are being explored as alternatives or complements to CAR T cells. Although significant durable responses are observed in relapsed/refractory B-cell malignancy patients treated with CAR T-cell therapies,9 severe adverse effects, including GVHD,14,39,40 CRS,21 and ICANS,20 have been clinical challenges. Conversely, many clinical studies have demonstrated that NK cell adoptive therapies are safe and effective, have a role in moderating graft-versus-leukemia responses while avoiding GVHD,41 and have resulted in minimal treatment-limiting toxicities.28 The underlying reason why NK cell therapy does not result in CRS or ICANS remains poorly understood. This may result from differences in NK persistence or expansion, the tight regulation by inhibitory receptors that prevent killing of healthy self tissues, or differential capacities to induce monocyte-derived cytokines that drive CRS (eg, IL-6).9,27,28,41 Li et al23 evaluated the CRS potential of iPSC-derived NK cells bearing CARs in NSG xenograft models and reported minimal toxicity.23 In human patients, CB-differentiated 19-CAR-NK cells exhibited no CRS,28 also consistent with an inherent biological difference between NK cells and T cells. Because the biology of ML NK cells between mice and humans is similar,25,42 immunocompetent mouse models may provide an ideal system to further elucidate the biology, safety, and efficacy of 19-CAR-ML NK cells in vivo in future studies.
NK cells have biological properties that provide a clear premise for investigation as cellular therapy. Because NK cells do not normally attack healthy tissues and do not express a clonally rearranged antigen receptor, they have limited capacity for causing GVHD. In addition, through their repertoire of activating receptors (eg, NKG2D, NKp46, NKp44, NKp30, DNAM-1), a CAR-engineered NK cell can still be activated via these receptors even if the tumor loses the CAR antigen, providing an inherent secondary recognition system. Furthermore, the normal expression of CD16 (FcγRIIIa) on mature NK cells allows for flexible dual-targeting strategies with therapeutic monoclonal antibodies or bispecific antibodies that ligate CD16 and a tumor-restricted antigen.43 Such dual targeting may also reduce target cell CAR antigen–based immune escape. NK cells have distinct trafficking patterns from T cells and circulate throughout the lymphoid systems and tissues.44 Thus, in situations where CAR T cells fail to localize to a tumor, CAR-ML NK cells may have unique access. Furthermore, CAR NK cell therapy can also be used in combined therapy with strategies to limit suppression, to fully unleash NK cell attack against tumor targets.45
There are a variety of distinct NK cellular therapeutics that are currently in preclinical or early-phase clinical development. These include genetically modified NK-92 immortalized cell lines,8,9 iPSCs differentiated into NK cells,46 accessory cell–expanded NKG2D-CAR NK cells,47 accessory particle–expanded NK cells,48 CB-differentiated 19-CAR NK cells,24 ML NK cells,10 and here CAR-ML NK cells. Each of these approaches is distinct and has strengths and potential limitations. For example, although extensive ex vivo expansion results in a large number of cells for flexible dose and schedule, there is a risk that the expanded NK cell populations may undergo senescence, depend on expansion signals for ongoing survival, or have altered homing.49 NK cells differentiated from early progenitors and precursors may have differences in signaling domains requirements23 or decreased maturation status and receptor profiles28 that could alter persistence and performance in vivo. Moreover, allogeneic NK cells are expected to be rejected by recipient immunity, unless engineered to reduce host T-cell recognition. Understanding the utility of each of these approaches will require testing in early-phase clinical trials. The results of the first-in-human clinical trial of CB-differentiated 19-CAR NK cells was recently reported and demonstrated 28-day clinical responses in 8 of 11 patients with relapsed/refractory B-cell lymphoma.28 The maximum dose of CB-differentiated CAR+ NK cells was well tolerated and was not associated with CRS, neurotoxicity, or GVHD, similar to prior NK cell adoptive immunotherapy studies.10,27 This initial study provides important evidence that 19-CAR–modified NK cells can safely induce remissions in relapsed/refractory B-cell malignancies.
We developed a distinct type of CAR-engineered NK cell. Starting with PB NK cells that were phenotypically and functionally mature, an ML program and CAR expression were simultaneously induced over a short 5-day culture period. ML NK cells were defined by an initial activation event (IL-12, IL-15, IL-18) followed by differentiation and a return to a resting state, with subsequent enhanced effector responses after restimulation with cytokines, activating receptors, or tumor targets.8,9,50 Previous work has established that ML NK cell biology improves on cNK cell antitumor properties, including enhanced functionality, activating receptor expression, longevity, and an expanded tumor recognition profile.51 We also established that adequate NK cell numbers may be isolated from a single donor leukapheresis to generate clinically useful ML NK cell products and that ML NK cells expand in vivo after adoptive transfer, within AML patients.10 Indeed, in an immune-compatible environment, ML NK cells persisted for >8 weeks after adoptive transfer.52 CAR-ML NK cells follow this same paradigm, where mature NK cells are briefly manipulated ex vivo, with full manifestation of those enhanced effector and expansion properties in vivo within the patient after adoptive transfer. Therefore, we expect that the in vivo expansion of CAR+ ML NK cells may also overcome prior limitations with viral vector–based engineering.53,54 This study provides important proof of principle that transduction efficiencies of 15% to 25% (lower compared with NK cell progenitor or iPSC transduction) are sufficient for clinical translation, and doses of ML NK cells obtained in AML clinical trials support the feasibility of generating adequate CAR-ML NK cells to treat patients. Finally, there remains an opportunity to further optimize CAR signaling for ML NK cells and other NK cell products with the use of constructs with NK cell–specific intracellular activating domains, including NKG2D and 2B4, because they have been shown to improve the function of CAR+ iPSC-differentiated NK cells.23 Overall, an optimal NK cell therapeutic will require multiple approaches to limit NK cell inhibition, augment antitumor function and persistence, and enhance tumor-specific recognition.
In summary, our study demonstrates the feasibility and proof of principle to generate CAR-modified ML NK cells from donor and patient PB. CAR-ML NK cells have an expanded antitumor recognition spectrum and exhibit specific and potent antitumor responses in vitro and in vivo. CAR-ML NK cells also have the potential to recognize hematological malignancies via different CAR specificities, vastly broadening the antitumor repertoire of PB NK cells. Thus, CAR-ML NK cells are a promising engineered NK cell approach to cellular cancer immunotherapy and warrant clinical testing in early-phase clinical trials.
Data may be shared after request to the corresponding author by e-mail (tfehnige@wustl.edu).
The online version of this article contains a data supplement.
The publication costs of this article were defrayed in part by page charge payment. Therefore, and solely to indicate this fact, this article is hereby marked “advertisement” in accordance with 18 USC section 1734.
Acknowledgments
The authors thank our patient volunteers and the lymphoma physician, nursing, and research assistant teams who care for them at the Washington University School of Medicine, acknowledge support from the Siteman Cancer Center Flow Cytometry Core (Bill Eades) and Immunomonitoring Laboratory (Stephen Oh), and thank Megan Cooper, Carl DeSelm, and Nathan Singh for insightful discussion.
This study was supported by a Medical Fellow Award from the Howard Hughes Medical Institute (M.G.); National Institutes of Health (NIH), National Cancer Institute (NCI) grants F32CA200253 (M.M.B.-E.), K12CA167540 (M.M.B.-E.), P50CA171063 (Specialized Programs of Research Excellence in Leukemia (M.M.B.-E., A.F.C., T.A.F.), and R01CA205239 (T.A.F.); NIH, National Heart, Lung and Blood Institute grant T32HL00708843 (P.W.); Siteman Cancer Center grant P30CA091842; the Leukemia and Lymphoma Society (T.A.F.); the V Foundation for Cancer Research (T.A.F.); the Children’s Discovery Institute at the Washington University School of Medicine (T.A.F.); the Jamie Erin Follicular Lymphoma Research Fund (T.A.F.); and the Steinback Fund (N.L.B.).
Authorship
Contribution: M.G., N.D.M., M.M.B.-E., and T.A.F. conceived and designed the study; M.G., N.D.M., P.W., M.M.B.-E., L.M., M.F., C.C.N., T.S., W.M., J.T., M.S., and F.G. collected, analyzed, and assembled the data; A.F.C., N.M.-S., N.L.B., and B.S.K. provided critical patient samples; M.D., M.Y.K., M.L.C., and J.F.D. provided critical reagents; M.G., N.D.M., M.M.B.-E., and T.A.F. wrote the manuscript; and all authors reviewed the data and edited and approved the final version of the manuscript.
Conflict-of-interest disclosure: Patents related to this study have been filed by Washington University (M.M.B.-E., T.A.F. inventors); N.M.-S. has served as a consultant for Kiowa Hakka Kirin and C4 Therapeutics and receives research funding from Bristol Myers-Squibb, Verastem Pharmaceuticals, Innate Pharmaceuticals, Genentech/Roche, Celgene, and Corvus Pharmaceuticals; M.L.C. has direct ownership of equity in and a consultancy with Wugen; J.F.D. is on the consulting/advisory committee for Rivervest, Bioline, Amphivena, Bluebird, Celegene, Incyte, NeoImuneTech, and Macrogenics and has ownership investment in Magenta and Wugen; T.A.F. has received research support from ImmunityBio, Compass Therapeutics, and HCW Biologics and advises Kiadis, Nkarta, Indapta, and Orca Biosystems. The remaining authors declare no competing financial interests.
Correspondence: Todd A. Fehniger, Washington University School of Medicine, 660 South Euclid Ave, Campus Box 8007, St. Louis, MO 63110; e-mail: tfehnige@wustl.edu.

This feature is available to Subscribers Only
Sign In or Create an Account Close Modal