

Key Points
Extended KRd plus transplant for NDMM patients provided high-quality responses with prolonged disease control and manageable tolerability.
Responses were rapid, but achieving best response required extended KRd treatment.

Abstract
In this phase 2 multicenter study, we evaluated the incorporation of autologous stem cell transplantation (ASCT) into a carfilzomib-lenalidomide-dexamethasone (KRd) regimen for patients with newly diagnosed multiple myeloma (NDMM). Transplant-eligible patients with NDMM received 4 cycles of KRd induction, ASCT, 4 cycles of KRd consolidation, and 10 cycles of KRd maintenance. The primary end point was rate of stringent complete response (sCR) after 8 cycles of KRd with a predefined threshold of ≥50% to support further study. Seventy-six patients were enrolled with a median age of 59 years (range, 40-76 years), and 35.5% had high-risk cytogenetics. The primary end point was met, with an sCR rate of 60% after 8 cycles. Depth of response improved over time. On intent-to-treat (ITT), the sCR rate reached 76%. The rate of minimal residual disease (MRD) negativity using modified ITT was 70% according to next-generation sequencing (<10−5 sensitivity). After median follow-up of 56 months, 5-year progression-free survival (PFS) and overall survival (OS) rates were 72% and 84% for ITT, 85% and 91% for MRD-negative patients, and 57% and 72% for patients with high-risk cytogenetics. For high-risk patients who were MRD negative, 5-year rates were 77% and 81%. Grade 3 to 4 adverse events included neutropenia (34%), lymphopenia (32%), infection (22%), and cardiac events (3%). There was no grade 3 to 4 peripheral neuropathy. Patients with NDMM treated with KRd with ASCT achieved high rates of sCR and MRD-negative disease at the end of KRd consolidation. Extended KRd maintenance after consolidation contributed to deepening of responses and likely to prolonged PFS and OS. Safety and tolerability were manageable. This trial was registered at www.clinicaltrials.gov as #NCT01816971.
Introduction
The treatment of multiple myeloma (MM) rapidly evolved with the introduction of immunomodulatory drugs (IMiDs) and proteasome inhibitors (PIs). The development of triplet combinations with these agents, clearly established as superior to doublets, has transformed the standard of care.1-4 Several triplet combinations are now used as induction therapy for patients with newly diagnosed MM (NDMM), including regimens with PI-chemotherapy or PI-IMiD backbones. PI-IMiD–based triplets have become a preferred treatment option because of their improved response and survival outcomes compared with previous standards of care.5
The combination of lenalidomide-bortezomib-dexamethasone (RVd) is one of the more commonly used first-line induction regimens and has shown consistently high activity across studies.2,6-8 The arrival of next-generation PIs brought additional PI-IMiD–based triplets to the clinic, including carfilzomib-lenalidomide-dexamethasone (KRd).9 RVd and KRd were the first regimens with 100% response rates, with some of the highest rates of complete response (CR) at that time.7,9 However, a preferred PI-IMiD–based regimen has not been clearly established in the first-line setting.10,11
Ongoing efforts to further improve treatment outcome in patients with NDMM include incorporation of monoclonal antibodies12-14 and/or autologous stem cell transplantation (ASCT) into treatment with novel regimens.14-18 Results from randomized trials have shown that incorporation of ASCT into RVd treatment improved clinical outcomes compared with RVd without ASCT, which supports the use of ASCT with novel regimens as a standard of care.17 In addition, there is emerging evidence that duration of treatment with multidrug regimens plays a role. Although the optimal duration of initial treatment is not well established, recent studies in transplant-ineligible patients indicate that extended treatment improves disease control compared with shorter, fixed durations, although a significant overall survival (OS) benefit has not been demonstrated.19-21 In studies of patients undergoing transplantation with multidrug induction and consolidation, the duration of triplet regimens such as RVd and KRd has generally been limited to 3 to 6 cycles of induction with 2 to 4 cycles of consolidation.7,8,17,22
Promising results have been reported with extended KRd (24 cycles) in patients with NDMM who were transplant ineligible, or they were eligible but they deferred ASCT.9 In this phase 2 multicenter Multiple Myeloma Research Consortium (MMRC) study, we show that incorporation of ASCT into a KRd regimen provided high rates of CR and stringent CR (sCR) after consolidation, with responses improving during extended KRd maintenance, likely contributing to observed prolonged progression-free survival (PFS) and OS outcomes.
Patients and methods
Study design and participants
This was a multicenter, open label, single-arm, phase 2 study. Patients were recruited from 5 MMRC sites in North America. Transplant-eligible patients age 18 years or older with NDMM who required systemic chemotherapy per International Myeloma Working Group (IMWG) uniform criteria were eligible.23 There was no upper age limit provided patients met transplant eligibility requirements. Full eligibility criteria are described in the supplemental Information, available on the Blood Web site. The study was conducted in accordance with US Food and Drug Administration and International Conference on Harmonization Guidelines for Good Clinical Practice, the Declaration of Helsinki, Health Canada, and any applicable health authority. The study protocol was approved by the institutional review boards or ethics committees of participating institutions. All patients provided written informed consent.
Treatment
Patients received 4 cycles (28 days per cycle) of KRd induction followed by ASCT, 4 cycles of KRd consolidation, and then 10 cycles of KRd maintenance, for a total of 18 cycles of KRd. For induction, carfilzomib was administered intravenously (IV) on days 1, 2, 8, 9, 15, and 16; the dose was 20 mg/m2 for days 1 and 2 of cycle 1 and then 36 mg/m2 thereafter. The carfilzomib dosing strategy was based on findings from the previous phase 1/2 study of KRd without transplantation.9 Lenalidomide was administered orally at 25 mg on days 1 to 21 of each cycle, and dexamethasone was given orally or IV at 40 mg on days 1, 8, 15, and 22 of each cycle. Dose modifications were permitted to manage toxicity (see supplemental Information for dose reduction guidance). The dosing and design of this study were similar to those of the previous KRd study without ASCT9 to help us better understand the potential impact of incorporating ASCT.
After KRd induction, granulocyte colony-stimulating factor and plerixafor were given for stem cell mobilization 2 to 4 weeks after the last dose of lenalidomide; alternative mobilization was allowed for treatment failures. Conditioning chemotherapy consisted of melphalan 200 mg/m2 IV. Participating centers followed their standard protocol for administering autologous peripheral stem cells, hydration, and prophylaxis measures.
KRd consolidation (cycles 5 to 8) was started within 70 to 90 days or <120 days after ASCT. Carfilzomib was administered on the same schedule as induction therapy at the last tolerated induction dose. Lenalidomide was restarted at 15 mg on days 1 to 21 for cycle 5 and then escalated to the last tolerated dose. Dexamethasone was administered at the same weekly dosing schedule as induction therapy but at a dose of 20 mg.
For KRd maintenance (cycles 9 to 18), carfilzomib was administered at the last tolerated consolidation dose on days 1, 2, 15, and 16. Lenalidomide and dexamethasone were administered at the last tolerated consolidation doses using the same dose schedules. After 18 KRd cycles, maintenance treatment was continued with single-agent lenalidomide at the last tolerated dose and schedule until disease progression or unacceptable toxicity.
Assessments
Disease-related tests were performed at baseline and on day 1 of each cycle, including M-protein by serum and/or urine protein electrophoresis, quantitative immunoglobulins, and immunoglobulin free light chains. Imaging was used according to the current IMWG recommendations, and response assessments were made according to IMWG uniform response criteria.24,25
Samples for minimal residual disease (MRD) analysis were obtained at landmark time points: after 8 cycles, after 18 cycles, and then at 1, 2, 3, and 5 years of single-agent lenalidomide maintenance. Samples were analyzed by next-generation sequencing at Adaptive Biotechnologies (Seattle, WA) with <10−5 sensitivity. MRD negativity was determined by IMWG criteria (patients with CR or better and with at least 10−5 MRD negativity).25 Positron emission tomography-computed tomography scans were performed at landmark time points in patients with MRD-negative status according to IMWG recommendations.
Adverse events (AEs) were graded according to the National Cancer Institute Common Terminology Criteria for Adverse Events version 4.0. AEs were collected from the day of treatment initiation through 30 days after the last dose or at the initiation of a new anticancer therapy. Per protocol, AEs were not collected during ASCT (stem cell collection through start of consolidation) and during single-agent lenalidomide maintenance.
Study end points
The primary end point was rate of sCR after 8 cycles of KRd. Secondary end points included overall response rate, PFS, and OS. Exploratory end points included evaluation of MRD rates at the indicated landmark time points.
Statistical analysis
For the primary end point, we estimated that 53 to 70 patients would be needed to interrogate the hypothesis that incorporation of ASCT into extended KRd would improve the sCR rate from 30% reported at the end of 8 cycles in the historical phase 1/2 study of KRd without ASCT9 to ≥50% after 8 cycles of KRd with ASCT. This study size provided 10% type II error (90% power) and a 5% type I error (2-sided) to detect an improvement of the sCR rate to 50% compared with the historical sCR rate of 30%.9
For the primary efficacy analysis, all patients were evaluated except those who declined to receive ASCT for reasons other than toxicity or efficacy. Patients who received <4 cycles of KRd consolidation were evaluable for the primary end point. For all other efficacy analyses, including time-to-event end points and safety, the results are based on the intent-to-treat (ITT) population, defined as all patients who received at least 1 dose of carfilzomib and lenalidomide. The MRD rate was estimated for the evaluable population and for a modified ITT (mITT) population as described by Perrot et al.26 For mITT, patients who failed to achieve a very good partial response (VGPR) or better or who did not complete KRd treatment for any reason were considered MRD positive, and patients who achieved a VGPR or better but did not have evaluable MRD were excluded from the analysis. To ensure adequate assessment of MRD, over-enrollment was allowed. Continuous and categorical data were summarized with descriptive statistics. Comparisons of categorical data were conducted with a Fisher’s exact test. Time-to-event end points were estimated by the Kaplan-Meier method with GraphPad Prism version 7.03 (La Jolla, CA). Statistical analyses were performed with SAS version 9.4 software.
Results
Between 29 January 2013, and 4 November 2015, 76 patients were enrolled and entered the induction phase. The data cutoff was 1 May 2019. Median age was 59 years (range, 40 to 76 years), with 28% age 65 years or older (Table 1). High-risk cytogenetic abnormalities according to IMWG criteria were detected in 36% of patients, including 15% with del(17p) and 11% with ultra-high-risk MM (≥3 cytogenetic abnormalities).27
Baseline characteristics
| Characteristic | n (%)* |
|---|---|
| Total no. of patients | 76 |
| Age, y | |
| Median (range) | 59 (40-76) |
| ≥65 | 21 (27.6) |
| Sex | |
| Male | 45 (59.2) |
| Female | 31 (40.8) |
| ECOG performance status | |
| 0-1 | 65 (85.5) |
| Unknown | 11 (14.5) |
| ISS stage | |
| I | 31 (40.8) |
| II | 31 (40.8) |
| III | 10 (13.2) |
| Unknown | 4 (5.3) |
| Cytogenetic risk by FISH† | |
| High | 27 (35.5) |
| del(17p) | 11 (14.5) |
| Ultra-high risk‡ | 8 (10.5) |
| Standard | 49 (64.5) |
| Serum β2-microglobulin, mg/L | |
| <3.5 | 45 (59.2) |
| ≥3.5 | 24 (31.6 |
| Unknown | 7 (9.2) |
| Characteristic | n (%)* |
|---|---|
| Total no. of patients | 76 |
| Age, y | |
| Median (range) | 59 (40-76) |
| ≥65 | 21 (27.6) |
| Sex | |
| Male | 45 (59.2) |
| Female | 31 (40.8) |
| ECOG performance status | |
| 0-1 | 65 (85.5) |
| Unknown | 11 (14.5) |
| ISS stage | |
| I | 31 (40.8) |
| II | 31 (40.8) |
| III | 10 (13.2) |
| Unknown | 4 (5.3) |
| Cytogenetic risk by FISH† | |
| High | 27 (35.5) |
| del(17p) | 11 (14.5) |
| Ultra-high risk‡ | 8 (10.5) |
| Standard | 49 (64.5) |
| Serum β2-microglobulin, mg/L | |
| <3.5 | 45 (59.2) |
| ≥3.5 | 24 (31.6 |
| Unknown | 7 (9.2) |
ECOG, Eastern Cooperative Oncology Group; FISH, fluorescence in situ hybridization; ISS, International Staging System.
All data in the table are n (%), unless otherwise designated.
Defined per International Myeloma Working Group (IMWG): t(4;14), del(17p), t(14;16), t(14;20), nonhyperdiploidy and gain(1q).
High, risk ≥3 cytogenetic abnormalities.27
Of the 76 patients, 64 completed 18 cycles of KRd (Figure 1). Median duration of KRd treatment was 18 cycles (range, 3 to 18 cycles). Twelve patients (16%) discontinued treatment early because of noncompliance (n = 1), patient/investigator preference (n = 3), disease progression (n = 4), and AEs (n = 4). Seventy-two patients underwent stem cell collection with a median yield of 8.2 × 106 cells/kg (range, 3.1-17.3 × 106 cells/kg), and 72 completed ASCT.
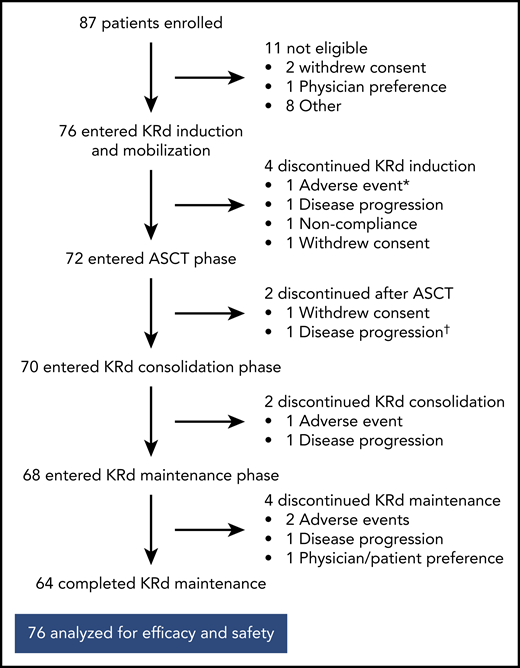
Patient disposition. *AE unrelated to treatment in patient retrospectively ineligible for the trial. †Discontinued on day +104 after ASCT and before starting KRd consolidation.
Patient disposition. *AE unrelated to treatment in patient retrospectively ineligible for the trial. †Discontinued on day +104 after ASCT and before starting KRd consolidation.
Efficacy
After 4 cycles, 97% of patients achieved a partial response or better, 73% a VGPR or better, 16% a CR or better, and 11% an sCR. After transplantation, the rate for CR or better was 25%, and it was 20% for sCR. At the primary end point (8 cycles of KRd), rates for CR or better and sCR increased to 65% and 60% and reached 79% and 76%, respectively, as best response in the ITT population (Figure 2; Table 2). Median time to sCR was 11.9 months (range, 0.9-26.4 months) (Figure 3).
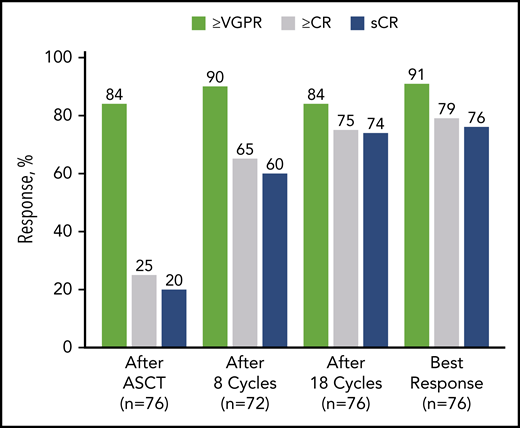
Response rates over the course of KRd plus ASCT treatment in ITT population (n = 76). After cycle 8 (n = 72), the per protocol population excluded 2 patients who withdrew consent, 1 patient for noncompliance, and 1 patient for AE unrelated to treatment. In the ITT population after cycle 8 (n = 76), the response rate for VGPR or better was 86%, for CR or better it was 62%, and for sCR it was 57%. nCR, near complete response; PR, partial response.
Response rates over the course of KRd plus ASCT treatment in ITT population (n = 76). After cycle 8 (n = 72), the per protocol population excluded 2 patients who withdrew consent, 1 patient for noncompliance, and 1 patient for AE unrelated to treatment. In the ITT population after cycle 8 (n = 76), the response rate for VGPR or better was 86%, for CR or better it was 62%, and for sCR it was 57%. nCR, near complete response; PR, partial response.
Best response
| Best overall response | KRd + ASCT, n (%) (n = 76) |
|---|---|
| sCR | 58 (76.3) |
| ≥CR | 60 (78.9) |
| ≥nCR | 67 (88.2) |
| ≥VGPR | 69 (90.8) |
| ≥PR | 74 (97.4) |
| Best overall response | KRd + ASCT, n (%) (n = 76) |
|---|---|
| sCR | 58 (76.3) |
| ≥CR | 60 (78.9) |
| ≥nCR | 67 (88.2) |
| ≥VGPR | 69 (90.8) |
| ≥PR | 74 (97.4) |
nCR, near complete response; PR, partial response.
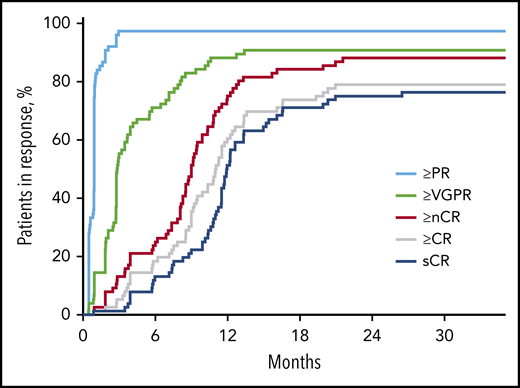
Time to response in respective response categories over time on treatment in months. nCR, near complete response; PR, partial response.
Time to response in respective response categories over time on treatment in months. nCR, near complete response; PR, partial response.
Among patients evaluated for MRD, the MRD-negative rate was 60% after 8 cycles, 70% after 18 cycles, and 81% as best response, with corresponding rates of 52%, 61%, and 70%, respectively, by mITT analysis (Figure 4). Sustained MRD-negative status per IMWG25 was observed in 55% of patients in the mITT population. Most patients (48 of 76) were continuing with lenalidomide maintenance at the cutoff date, and of those completing the last landmark MRD evaluation at 3 years of lenalidomide maintenance, 76% (22 of 29) were MRD negative.
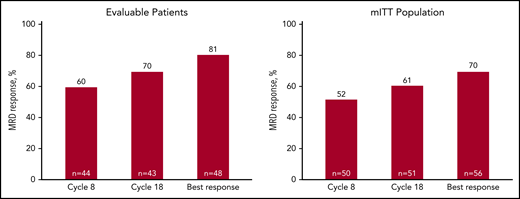
MRD response rates over the course of KRd plus ASCT treatment. Forty-eight patients had baseline and at least 1 postbaseline bone marrow sample available for MRD assessment by next-generation sequencing (clonoSeq). MRD rates at 8 cycles, at the end of 18 cycles of KRd, and best response in evaluable patients (patients with at least 1 MRD assessment) (A), and in mITT population (B) (as described by Perrot et al26 and in “Methods”).
MRD response rates over the course of KRd plus ASCT treatment. Forty-eight patients had baseline and at least 1 postbaseline bone marrow sample available for MRD assessment by next-generation sequencing (clonoSeq). MRD rates at 8 cycles, at the end of 18 cycles of KRd, and best response in evaluable patients (patients with at least 1 MRD assessment) (A), and in mITT population (B) (as described by Perrot et al26 and in “Methods”).
After a median follow-up of 56.0 months (range, 2.9-75.1 months), median PFS and OS were not reached. The estimated 5-year PFS rate was 72% (95% CI, 60%-81%), and the 5-year OS rate was 84% (95% CI, 71%-92%) (Figure 5A-B). Among 39 patients who were MRD negative, estimated 5-year PFS was 85% (95% CI, 69%-93%), and OS was 91% (95% CI, 75%-97%) (Figure 5C-D).
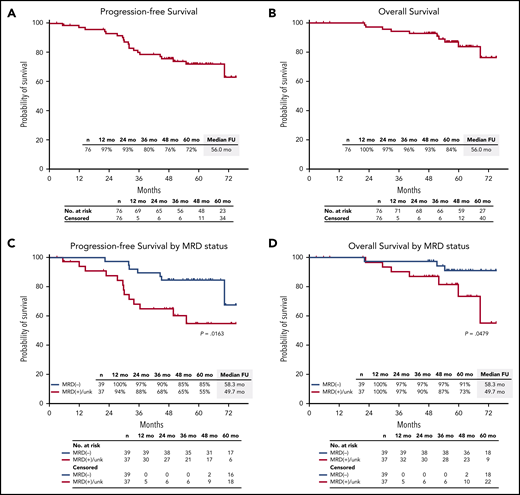
PFS and OS in the ITT population. PFS and OS in the ITT population (A-B), and by MRD status (C-D). FU, follow-up; unk, unknown.
PFS and OS in the ITT population. PFS and OS in the ITT population (A-B), and by MRD status (C-D). FU, follow-up; unk, unknown.
Response rates were not statistically different by IMWG risk status, with sCR as best response in 81% of patients (22 of 27) with high-risk cytogenetics and 73% of patients (36 of 49) considered standard risk (P = .58). In the mITT population, MRD negativity was achieved by 72% of high-risk patients (13 of 18) and 68% of standard-risk patients (26 of 38) at data cutoff. Among 11 patients with del(17p), 8 achieved sCR, and 2 of 4 patients who were evaluable achieved an MRD-negative response. The 5-year PFS rate was 57% for high-risk and 81% for standard-risk patients, and the 5-year OS rates were 72% and 92%, respectively (Figure 6A-B). For patients with high-risk cytogenetics, MRD negativity was associated with improved PFS compared with MRD-positive/MRD-unknown patients (P = .04) with no significant difference in OS (P = .26) (Figure 6C-D). For patients who achieved MRD negativity, there was no statistically significant difference between those with high-risk cytogenetics and those who had standard-risk cytogenetics for PFS (P = .31) or for OS (P = .13).
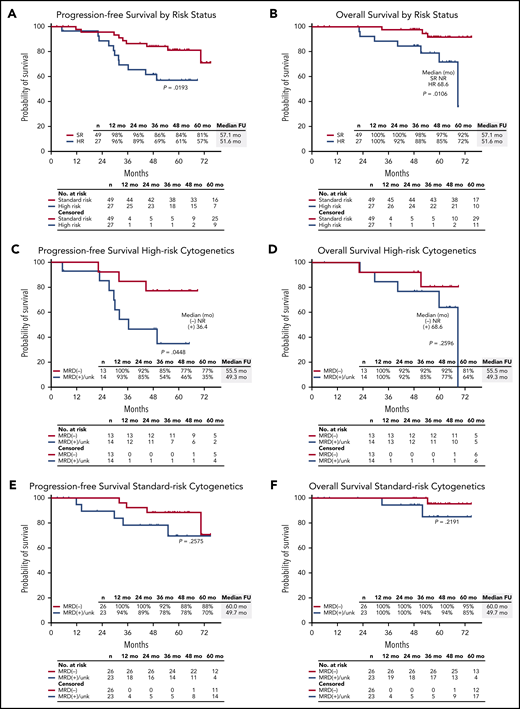
PFS and OS in the ITT population. PFS and OS in the ITT population by (A-B) high-risk (HR) vs standard-risk (SR) cytogenetics, (C-D) in patients with high-risk cytogenetics by MRD status by next-generation sequencing, and (E-F) in patients with standard-risk cytogenetics by MRD status by next-generation sequencing. NR, not reached.
PFS and OS in the ITT population. PFS and OS in the ITT population by (A-B) high-risk (HR) vs standard-risk (SR) cytogenetics, (C-D) in patients with high-risk cytogenetics by MRD status by next-generation sequencing, and (E-F) in patients with standard-risk cytogenetics by MRD status by next-generation sequencing. NR, not reached.
Safety and tolerability
KRd treatment was well tolerated during the study. AEs were manageable with dose modifications, with a rate of 76% for carfilzomib, 66% for dexamethasone, and 71% for lenalidomide. Four patients discontinued treatment because of AEs.
The most common AEs (all grades) were infection (74%), fatigue (67%), and thrombocytopenia (62%) (Table 3). The most common grade 3 to 4 AEs were neutropenia (34%), lymphopenia (32%), and infection (22%). Hypertension (all grades) was reported in 20% of patients, with 5% experiencing grade 3 to 4 hypertension. Forty-two percent of patients developed grade 1 to 2 peripheral neuropathy (7% grade 2); there were no grade 3 to 4 events. Cardiac events were infrequent (13%) and generally moderate in severity (grade 1 to 2). Two patients experienced asymptomatic decline of left ventricular ejection fraction to 45% to 50% pre-transplant, and 1 patient experienced a transient decline to 47% associated with hypertension during KRd maintenance. There was 1 case of secondary primary malignancy (acute myeloid leukemia) during the study, and one case of thrombotic microangiopathy (thrombotic thrombocytopenic purpura/hemolytic uremic syndrome), which developed within 1 year of KRd completion and during lenalidomide maintenance. There were no treatment-related deaths.
Treatment-emergent AEs during KRd*
| AE | KRd + ASCT (n = 76) | |
|---|---|---|
| All grades, n (%) | Grade 3 to 4, n (%) | |
| Hematologic | ||
| Thrombocytopenia | 47 (62) | 11 (14) |
| Anemia | 32 (42) | 9 (12) |
| Lymphopenia | 32 (42) | 24 (32) |
| Neutropenia | 30 (39) | 26 (34) |
| Nonhematologic | ||
| Infection | 56 (74) | 17 (22) |
| Fatigue | 51 (67) | 4 (5) |
| Diarrhea | 39 (51) | 7 (9) |
| Hyperglycemia | 33 (43) | 6 (8) |
| Dyspnoea | 30 (39) | 2 (3) |
| Peripheral neuropathy | 32 (42) | 0 |
| Rash | 33 (43) | 4 (5) |
| Hypophosphatemia | 22 (29) | 11 (14) |
| Hypertension | 15 (20) | 4 (5) |
| Thromboembolic events | 14 (18) | 5 (7) |
| Cardiac events† | 10 (13) | 2 (3) |
| AE | KRd + ASCT (n = 76) | |
|---|---|---|
| All grades, n (%) | Grade 3 to 4, n (%) | |
| Hematologic | ||
| Thrombocytopenia | 47 (62) | 11 (14) |
| Anemia | 32 (42) | 9 (12) |
| Lymphopenia | 32 (42) | 24 (32) |
| Neutropenia | 30 (39) | 26 (34) |
| Nonhematologic | ||
| Infection | 56 (74) | 17 (22) |
| Fatigue | 51 (67) | 4 (5) |
| Diarrhea | 39 (51) | 7 (9) |
| Hyperglycemia | 33 (43) | 6 (8) |
| Dyspnoea | 30 (39) | 2 (3) |
| Peripheral neuropathy | 32 (42) | 0 |
| Rash | 33 (43) | 4 (5) |
| Hypophosphatemia | 22 (29) | 11 (14) |
| Hypertension | 15 (20) | 4 (5) |
| Thromboembolic events | 14 (18) | 5 (7) |
| Cardiac events† | 10 (13) | 2 (3) |
AEs with rate of >10% for any grade; events during ASCT and single-agent lenalidomide maintenance were not captured per protocol.
Two patients had asymptomatic left ventricular ejection fraction of 45% to 50% pretransplant, and one had a transient decline of left ventricular ejection fraction to 47% associated with hypertension during KRd maintenance.
Discussion
In this single-arm, phase 2 study, patients with NDMM receiving KRd with ASCT showed high rates of deep, durable responses. After 8 cycles of KRd with ASCT, 65% of patients achieved CR or better and 60% achieved sCR (primary end point). Extended KRd maintenance after consolidation further improved depths of response, with CR and sCR rates improving to 79% and 76% as best response by ITT. High CR and sCR rates corresponded to high MRD-negative rates, 70% by next-generation sequencing at <10−5 sensitivity in the mITT population. These response rates seem to exceed rates reported in studies of KRd without transplantation9 and are among the highest rates in NDMM, including those reported in recent studies with triplet combinations plus monoclonal antibodies with or without ASCT.12-14,18
After prolonged follow-up (median, 5 years), median PFS and OS have still not been reached, with close to 70% of patients free from progression at 5 years. For patients who achieved an MRD-negative response, the 5-year PFS was 85%, which was significantly higher than the PFS rate of 55% for MRD-positive/MRD-unknown patients. The 5-year PFS rates of both MRD-negative and MRD-positive/MRD-unknown patients seem higher than rates reported in previous studies with comparable follow-up.17 This may reflect the depth of response across all enrolled patients, with a rate for VGPR or better of 91%, indicating that regardless of MRD status, extended KRd with ASCT is very effective.
The initial response to KRd was rapid, and responses continued to improve beyond 12 months of treatment for many patients, indicating that extended treatment with highly active multidrug regimens are needed to achieve quality durable responses across a spectrum of patients in an NDMM population. sCR responses continued to increase beyond 8 cycles of treatment, with a median time to sCR of 11.9 months; 42% of conversions to sCR occurred 12 months after the start of KRd treatment, with an upper range of 20 months. Although MRD conversion was not assessed for each treatment cycle, MRD-negative rates also increased at landmark time points; the increase between cycle 8 and 18 was relatively modest and may indicate that MRD-negative status in some patients can be reached earlier than the clearance of M-protein. This reflects the predictive significance of MRD and the established phenomenon of slower clearance of M-protein than clonal plasma cells in bone marrow. Overall, our results indicate that KRd treatment beyond 8 cycles has the potential for deepening responses, possibly contributing to the observed prolonged PFS at 5 years in the ITT population and for both MRD-negative and MRD-positive/MRD-unknown patients.
The primary objective of the study was to evaluate the addition of ASCT to KRd by using sCR rate at the end of KRd consolidation (total 8 KRd cycles) as the surrogate end point of efficacy. The sCR rate of 60% after a total of 8 cycles of KRd, exceeded the predefined threshold of 50%, which was based on the sCR rate of 30% after 8 cycles in a previous KRd study without ASCT.9 In addition, extending treatment with KRd maintenance seemed to improve multiple secondary end points compared with extended KRd without ASCT,9 including higher rates of sCR as best response and MRD-negative disease, although there are important limitations with cross-study comparisons. In all, results of this study support the premise that incorporating ASCT into extended KRd can improve clinical outcomes, which supports the need for further evaluation in the randomized setting.
The phase 2 FORTE trial randomly assigned patients with NDMM to 8 cycles of KRd with ASCT, 12 cycles of KRd without ASCT, or 8 cycles of carfilzomib-cyclophosphamide-dexamethasone (KCd) with ASCT, with a second random assignment to KR or lenalidomide maintenance.15,28,29 Preliminary results demonstrated that both KRd regimens outperformed KCd with respect to sCR after consolidation, but there was no difference between the KRd arms, which would seem to contradict our premise that the addition of ASCT improves outcomes. In the FORTE study, the rates for CR or sCR or better were 60% and 44% in KRd with ASCT after 8 cycles vs 61% and 43% in KRd without ASCT after 12 cycles; the MRD-negative response rate was 58% vs 54%, respectively.28 However, with the landmark evaluation of KRd without ASCT at 12 cycles in the FORTE trial, higher rates of CR or sCR or better and MRD are expected compared with 8 cycles. An early analysis of the FORTE study showed trends in response at landmark time points consistent with this study: the rates of CR or better or sCR for KRd with ASCT increased from 28% and 20% after ASCT to 50% and 41% after 8 cycles, whereas in the KRd without ASCT arm, the sCR rates increased from 35% and 31% after 8 cycles to 52% and 42% after 12 cycles.15 We designed this study to compare the sCR rate after 8 cycles of KRd with ASCT with the sCR rate after 8 cycles from the historical study of KRd without ASCT.9 In addition, a recent update from the FORTE trial showed a lower risk of early relapse for KRd with ASCT compared with KRd without ASCT and a higher rate of sustained MRD negativity, suggesting that the addition of ASCT may improve treatment outcomes with KRd,29,30 similar to results from the IFM2009 trial of RVd with and without ASCT.17 Follow-up for PFS and OS is ongoing for the FORTE trial and will help to differentiate between KRd with and without ASCT.
To understand the potential benefit of extended KRd with ASCT, toxicity risks also need to be carefully considered. Overall, KRd with ASCT was well tolerated during induction and through consolidation and maintenance. Treatment toxicity was managed with dose modification, and discontinuations were infrequent. The types and rates of AEs in this study were consistent with those from KRd without ASCT9 and with results from the FORTE trial15 ; cytopenias and infections were the most common grade 3 to 4 events. Peripheral neuropathy was reported in almost half the patients, but most events were grade 1 in severity, with no grade 3 to 4 events. In a phase 2 study, the IFM group reported 8 serious cardiovascular events in 44 patients with NDMM who received KRd with ASCT.22 In the current study and the FORTE trial,15 cardiovascular events were present but manageable. We need to better understand long-term risk with KRd with ASCT in a broader patient population, and the safety results from ongoing randomized trials with KRd should help resolve these issues. Recently, the phase 3 ENDURANCE study, which compared KRd with RVd in patients with NDMM not undergoing ASCT, reported a higher rate of cardio-pulmonary-renal events with KRd but a higher rate of peripheral neuropathy with RVd.11 The ongoing phase 3 ATLAS trial (NCT02659293) will compare the efficacy and safety of maintenance therapy with KRd vs lenalidomide after transplantation.
In this study, the best response rates by cytogenetic risk, including sCR rates, were comparable, but PFS curves for high-risk and standard-risk patients diverged after 24 months, reflecting differences in durability of response and resulting in a shorter median PFS for the high-risk subgroup. However, PFS outcomes for high-risk patients who achieved MRD-negative response (∼50% of all high-risk patients [n = 27]) were markedly improved compared with MRD-positive/MRD-unknown high-risk patients and compared with standard-risk patients. This observation is consistent with previous observations from the GEM/PETHEMA Study Group which showed MRD response was more relevant than CR in overcoming the negative prognostic impact of high-risk cytogenetics.31 Considering the relatively high rates of MRD negativity for high-risk patients in this study, our results support further evaluation of extended KRd with ASCT in high-risk patients, with or without the addition of novel agents to achieve MRD-negativity.6,27,32,33
Several recent studies in NDMM have shown a clear benefit of adding antibodies (eg, daratumumab) to doublet and triplet regimens with and without ASCT.12-14,18 In the phase 3 CASSIOPEIA study, the addition of daratumumab to bortezomib, thalidomide, and dexamethasone (VTd) induction/consolidation in patients with NDMM undergoing ASCT significantly improved the sCR rate 100 days after ASCT to 29% compared with 20% for patients who received VTd alone.14 Similarly, the addition of daratumumab to RVd induction/consolidation (D-RVd) for patients with NDMM undergoing ASCT in the phase 2 GRIFFIN study improved the sCR rate at the end of consolidation to 42.4% compared with 32.0% for RVd alone.18 Patients in the D-RVd arm received D-R maintenance after consolidation whereas those in the RVd arm received lenalidomide maintenance; after a median follow-up of 22.1 months, the sCR rate was 62.6% with D-RVd compared with 45.4% with RVd, and the MRD-negative rate (<10−5 sensitivity) was 51.0% compared with 20.4%.
Our results support strategies to further enhance active regimens, with the objective of improving sCR and MRD-negative rates, particularly for patients with high-risk disease. Whether ASCT or an antibody added to a backbone of existing regimens, or both, would ultimately be preferred will need to be sorted out and may include response-adaptive strategies based on achievement of MRD negativity and more active maintenance regimens. Given the improvement in MRD response with daratumumab in the GRIFFIN study,18 its addition to KRd is of high interest, and preliminary results of KRd-daratumumab regimens in NDMM have reported very high MRD-negative rates (80% to 83% at <10−5 sensitivity).16,34,35
Moving forward, it will be important to better define which multidrug regimens to use as extended treatment backbones and in which patients. Several studies have assessed RVd with ASCT. In the phase 3 IFM2009 study, patients who received RVd-ASCT had longer PFS compared with those who received RVd (50 vs 36 months), and a higher CR rate as best response (59% vs 48%).17 In a second phase 3 study, PETHEMA/GEM2012, patients were randomly assigned to 2 different ASCT conditioning regimens with all patients receiving RVd induction/consolidation; in a grouped response analysis, the sCR rate was 44.5% after consolidation, with a corresponding MRD-negative rate (<10−6 sensitivity) of 45.2%.8
The results reported here and from other KRd studies9,15,22,36,37 support KRd for patients undergoing ASCT, but definitive studies are needed to determine its role, particularly in relation to RVd, in the current treatment landscape. The phase 3 ENDURANCE study compared KRd and RVd regimens in patients with NDMM, with no intention for immediate ASCT and without high-risk disease.11 Patients were randomly assigned to induction with KRd (9 cycles, 4 weeks per cycle) or RVd (12 cycles, 3 weeks per cycle), with a second random assignment to indefinite or 2 years of lenalidomide maintenance. Initial outcomes were recently presented, with no significant difference in the co-primary end point of PFS (median, 34.6 months for KRd vs 34.4 months for RVd), although KRd was associated with a higher rate of VGPR or better after induction (73.8% vs 64.7%). Although these results support the view that there is no significant difference between KRd and RVd in NDMM, outcomes seem inferior to results from other KRd studies to date, including in the non-transplantation arm of the FORTE trial,9,15,36,37 as well as RVd studies, including large randomized trials.2,7,17 At this point, it is unclear whether the inconsistency of outcomes from the ENDURANCE trial with historical studies may be related to differences in study design (eg, phase 2 vs phase 3), study populations, or relatively short durations of treatment in both arms, a potential key driver of efficacy with KRd in the current study. The ongoing randomized phase 3 COBRA study (NCT03729804) is comparing extended KRd (24 cycles)9 with the established RVd regimen (8 cycles with lenalidomide maintenance),2,17 both arms allowing high-risk patients and candidates for transplantation but with deferred ASCT. More recently, investigators initiated the phase 2 ADVANCE study (NCT04268498), which will randomly assign patients with NDMM to RVd, KRd, or D-KRd. Taken together, these studies, as well as the ongoing FORTE and ATLAS trials, should help to better define a preferred regimen in NDMM and for which patient population, and which regimen or strategy will be more suitable for making further progress toward elimination of the disease.
In the era of highly active triplet and now quadruplet combinations, differentiating between treatment regimens in general NDMM populations with standard survival end points may prove to be increasingly challenging, which highlights the need for surrogate markers. MRD is emerging as a likely surrogate candidate, because multiple studies have demonstrated strong association between MRD status and survival outcomes.8,14,17,18 Several ongoing randomized trials, including the ADVANCE study, have already adopted MRD as a primary end point, and regulatory agencies are considering MRD as a surrogate end point. There are also efforts to adopt risk- and MRD-adaptive treatment strategies for intensification and/or extending treatment duration as well as de-escalation or discontinuation.16,38,39 MRD-negative response, or preferably sustained MRD-negative response, could be used to inform treatment of standard-risk patients in whom extended multidrug regimens or even ASCT may not be of benefit, or conversely, for high-risk patients who may require longer multidrug treatment to achieve sustained MRD response or the addition of novel agents.31 Whether ASCT or an antibody, or both, should be added to a backbone of existing regimens along with duration of treatment will ultimately need to be sorted out in ongoing and future prospective randomized trials.
In summary, results from the phase 2 study reported here demonstrate that patients treated with extended KRd with ASCT achieved high rates of sCR and MRD-negative disease with prolonged PFS and OS. The sCR rate after KRd consolidation met the primary end point, with responses continuing to improve during KRd maintenance. In addition, we observed that KRd with ASCT generates high rates of MRD negativity in both high-risk and standard-risk patients, and that the achievement of MRD-negative response in high-risk patients is associated with improved outcomes, comparable to those of standard-risk patients. Safety and tolerability were acceptable and manageable. Overall, results from this study, together with those of other KRd studies,9,22,36 support ongoing and future evaluations of extended KRd with and without ASCT in the randomized setting.
Presented in abstract form at the 59th Annual Meeting and Exposition of the American Society of Hematology, Atlanta, GA, 9-12 December 2017.
For further information, please contact the corresponding author.
The online version of this article contains a data supplement.
The publication costs of this article were defrayed in part by page charge payment. Therefore, and solely to indicate this fact, this article is hereby marked “advertisement” in accordance with 18 USC section 1734.
Acknowledgments
The authors thank Daniel Juergens, Amanda McIver, David Johnson, Can Rui Jiang, Shaun Rosebeck, Fabrizio Fazzi, Agata Turowski, Bernadette Libao, Evangelia Andreatos, Megan Whelan, Christine Gleason, and Becky Malloy at the University of Chicago for their assistance in conducting this study.
This study was supported, in part, by Amgen, Celgene (a Bristol-Myers Squibb company), and the Multiple Myeloma Research Consortium. Study funding supported medical writing and editorial assistance by Michael Raffin (Fishawack Communications, Conshohocken, PA).
Authorship
Contribution: J.K.J., N.R., R.V., D.R., J.B., K.A.G., T.M.Z., and A.J.J. conceived and designed the trial; J.K.J., T.K., N.R., R.V., D.R., J.B., B.A.D., C.A.R., P.R., S.G., A.T.S., L.S., K.M.T., T.H., A.E.R., D.D., K.A.G., and A.J.J. analyzed and interpreted the data; J.K.J., T.K., R.V., D.R., J.B., B.A.D., S.G., A.T.S., D.D., K.A.G., T.M.Z., and A.J.J. drafted the manuscript; and all authors collected and assembled the data and critically reviewed and approved the final manuscript.
Conflict-of-interest disclosure: N.R. had a consulting or advisory role with Amgen, Bristol-Myers Squibb, Celgene, Janssen Oncology, Merck, Novartis, and Takeda and received research funding from AstraZeneca. R.V. received honoraria from, had a consulting or advisory role with, and received funding for travel, accommodations, and expenses from Bristol-Myers Squibb and Celgene. D.R. received honoraria from Amgen, Celgene, Janssen, and Takeda, had a consulting or advisory role with Amgen, Celgene, Janssen, Karyopharm Therapeutics, and Takeda, received research funding from Bristol-Myers Squibb, Celgene, Janssen, Merck, Otsuka, and Takeda, and provided expert testimony for Amgen and Celgene. J.B. had a consulting or advisory role with Amgen, BioClinica, Bristol-Myers Squibb, Celgene, Crispr Therapeutics, Janssen, Karyopharm Therapeutics, Kite Pharma, Legend Biotech, Prothena, Servier, and Takeda, and received research funding from AbbVie, Acetylon, Amgen, Bluebird Bio, Bristol-Myers Squibb, Celgene, Constellation, CURIS, Genentech/Roche, Glenmark, Janssen, Kesios, Eli Lilly, Novartis, Poseida Therapeutics, Sanofi, Takeda, Teva, and Vivolux. P.R. had a consulting or advisory role with Celgene, Janssen, Jazz Pharmaceuticals, Karyopharm Therapeutics, Oncopeptides, Sanofi, and Takeda and received research funding from Bristol-Myers Squibb, Celgene, Oncopeptides, and Takeda. D.D. received honoraria and research funding from Amgen and Celgene. T.M.Z. received honoraria from, had a consulting or advisory role with, and served on the speakers’ bureau for Celgene and Amgen. A.J.J. received honoraria from Amgen, Bristol-Myers Squibb, Celgene, and GlaxoSmithKline, was an investigator for AbbVie-sponsored clinical trials, Janssen, Karyopharm Therapeutics, Millennium Pharmaceuticals, Sanofi, Skyline Diagnostics, and Takeda, and had a consulting or advisory role with Amgen, Bristol-Myers Squibb, Celgene, and GlaxoSmithKline. The remaining authors declare no competing financial interests.
The current affiliation for T.K. is Poznan University of Medical Sciences, Poznan, Poland.
The current affiliation for C.A.R. is Weill Cornell Medicine, New York, NY.
The current affiliation for T.M.Z. is BeiGene, San Mateo, CA.
Correspondence: Andrzej J. Jakubowiak, Section of Hematology/Oncology, University of Chicago Medical Center, 5841 S Maryland Ave, MC 2115, Chicago, IL 60637-6613; e-mail: ajakubowiak@medicine.bsd.uchicago.edu.
