

Key Points
This study reveals the network of allosteric and factor VIII-driven changes in activated FIX associated with catalytic rate enhancement.
Disruption of this network explains why single mutations in FIX may silence the response to FVIII and thereby cause hemophilia B.

Abstract
The assembly of the enzyme-activated factor IX (FIXa) with its cofactor, activated factor VIII (FVIIIa) is a crucial event in the coagulation cascade. The absence or dysfunction of either enzyme or cofactor severely compromises hemostasis and causes hemophilia. FIXa is a notoriously inefficient enzyme that needs FVIIIa to drive its hemostatic potential, by a mechanism that has remained largely elusive to date. In this study, we employed hydrogen–deuterium exchange-mass spectrometry (HDX-MS) to investigate how FIXa responds to assembly with FVIIIa in the presence of phospholipids. This revealed a complex pattern of changes that partially overlaps with those changes that occur upon occupation of the substrate-binding site by an active site-directed inhibitor. Among the changes driven by both cofactor and substrate, HDX-MS highlighted several surface loops that have been implicated in allosteric networks in related coagulation enzymes. Inspection of FVIIIa-specific changes indicated that 3 helices are involved in FIXa–FVIIIa assembly. These are part of a basic interface that is also known as exosite II. Mutagenesis of basic residues herein, followed by functional studies, identified this interface as an extended FVIIIa-interactive patch. HDX-MS was also applied to recombinant FIXa variants that are associated with severe hemophilia B. This revealed that single amino acid substitutions can silence the extended network of FVIIIa-driven allosteric changes. We conclude that HDX-MS has the potential to visualize the functional impact of disease-associated mutations on enzyme–cofactor complexes in the hemostatic system.
Introduction
Upon vessel injury, the coagulation cascade is activated, which, in concert with platelets and vascular components, ultimately leads to fibrin clot formation and hemostasis. One key constituent of the coagulation cascade is the complex of factor IX (FIX) and factor VIII (FVIII). Qualitative or quantitative defects of FVIII or FIX are associated with the bleeding disorders hemophilia A or B, respectively.1 FIX belongs to the chymotrypsin-like serine proteases.2 These circulate as inactive precursors (zymogens) that need proteolytic activation to develop enzymatic activity, and usually require a cofactor to reach full catalytic potential.2 The zymogen FIX is a single-chain protein that is converted into activated FIX (FIXa) by activated factor VII (FVIIa) or activated factor XI.3 FIXa comprises a light chain with an N-terminal domain rich in γ-carboxyglutamic acid (the Gla domain), 2 epidermal growth factor (EGF)–like domains (EGF-1 and EGF-2), and a heavy chain that represents the C-terminal protease domain.3-5 Like its homolog FVIIa, FIXa displays low intrinsic catalytic potential in the absence of cofactors.2,6 Full FIXa activity requires assembly with activated FVIII (FVIIIa) in the presence of Ca2+ ions and phospholipids.7 FVIIIa is the activated derivative of the procofactor FVIII, a large heterodimeric glycoprotein composed of a heavy chain (domains A1-A2-B) and a light chain (domains A3-C1-C2).8 Proteolytic cleavage at domain junctions yields FVIIIa, a trimer of the segments A1 and A3-C1-C2 and the loosely associated A2 domain, thus making FVIIIa a labile component with transient cofactor activity.8 When assembled in the FIXa-FVIIIa complex, FVIIIa enhances FIXa activity by more than 104-fold, thus driving the conversion of factor X (FX) into activated FX (FXa) to physiologically relevant levels.6
During the past decades, the identification of molecular variants of FVIII and FIX in hemophilia A and B patients have guided biochemical studies employing site-directed mutagenesis and functional characterization to obtain deeper understanding of FIXa and FVIIIa structure and function. As for the FIXa–FVIIIa complex, putative interactive sites have been inferred mostly by combining functional studies with modeling and crystallography of its constituents or homologs thereof.3,4,7 For instance, the EGF-1/EGF-2 interface in the FIXa light chain has proven important for FVIIIa-driven rate enhancement and interaction with the FVIIIa light chain.9,10 As for the FIXa protease domain, the 330-helix{162CT} (CT subscript indicates chymotrypsinogen numbering) seems to interact with the 557-565 loop on the A2 domain of FVIIIa, whereas residues 301-303{132-134CT} may bind FVIIIa residues 698-712.11-13
Full understanding of FIXa–FVIIIa complex assembly has not been accomplished so far. A complete crystal structure of FIXa, including the lipid-binding Gla domain, is still lacking, and most of the available human FIXa crystals are limited to truncated EGF-2/protease domain constructs.4,5,14,15 Cocrystallization of FIXa with FVIIIa has been hampered by the inherent instability of the FVIIIa trimer. Another challenge is the need for phospholipids, which are normally supporting assembly of the FIXa–FVIIIa complex.6,7,16 As a consequence, structural information is still insufficient to understand the molecular mechanism of FVIIIa-driven rate enhancement of FIXa.
Hydrogen–deuterium exchange mass spectrometry (HDX-MS) is increasingly used for the study of protein complexes and dynamics. In the field of blood coagulation it has been successfully used to establish the footprint of the interaction between, for instance, thrombomodulin and thrombin,17 and FVIII with von Willebrand factor,18,19 phospholipid membranes,20 and anti-FVIII antibodies.21,22 In addition, HDX-MS has been used to unravel allosteric networks within thrombin23-25 and FVIIa.26
This study aimed to explore cofactor- and substrate-driven events in FIXa in the presence of phospholipids by HDX-MS. Binding and functional studies in combination with HDX-MS of recombinant dysfunctional FIXa variants were further employed to assess the functional implications thereof. This research provides a comprehensive view on the ensemble of cofactor-mediated structural changes and allosteric crosstalk within the FIX molecule.
Materials and methods
Materials
The QuikChange Kit was from Agilent Technologies (Middelburg, The Netherlands). Glu-Gly-Arg-chloromethylketone (EGRck) was from Bachem (Bubendorf, Switzerland). Factor XIa was from Enzyme Research Laboratories (South Bend, IN). Calcium chloride solution (1 M) and 99.9% deuterium oxide were from Sigma-Aldrich (St Louis, MO). Ultrapure urea, molecular biology–grade 5 M NaCl solution and Tris-HCl were from Invitrogen (Breda, The Netherlands). The chicken egg L-α-phosphatidylcholine (PC) and porcine brain L-α-phosphatidylserine (PS) were from Avanti Polar Lipids Inc (Alabaster, AL). NaCl was from Fagron (Rotterdam, The Netherlands), and N-2-hydroxyethylpiperazine-N′-2-ethanesulfonic acid (HEPES) was from Serva (Heidelberg, Germany). Human serum albumin (HSA) was obtained from the Division of Products of Sanquin (Amsterdam, The Netherlands). Tris(2-carboxyethyl)phosphine hydrochloride (TCEP-HCl) was obtained from Thermo Fisher Scientific (Breda, The Netherlands). HEPES buffer solution (1 M) was from Gibco (Paisley, United Kingdom). All other chemicals were from Merck (Darmstadt, Germany).
FIX and FIXa variants
FIX variants with mutations E78K, K293A{126CT}, R333A{165CT}, R338{170CT}, K341{173CT}, K400A{230CT}, R403A{233CT}, and FIXaR333A+R403A{165+233CT} were obtained by site-directed mutagenesis in a pcDNA3.1(-) vector encoding wild-type FIX27 with the QuikChange Kit using appropriate primers. Mutagenesis was confirmed with the complete sequencing of the FIX encoding parts on the mutated plasmid, using BigDye Terminator Sequencing Kit (Applied Biosystem, Foster City, CA). Recombinant FIX and FIX variants were expressed in mammalian cells as described elsewhere.27,28 Recombinant FIX was immunopurified and converted into FIXa by FXIa-mediated limited proteolysis as previously described27,29 and stored in 20 mM HEPES (pH 7.4), 150 mM NaCl, and 50% (volume-to-volume ratio [v/v]) glycerol at −20°C. FIXaEGR was prepared by incubation of FIXa (4 μM) with EGRck (10 mM) for 45 minutes at 37°C in a buffer containing 20 mM HEPES (pH 7.4), 150 mM NaCl, and 10 mM CaCl2. Unbound EGRck was removed by dialysis initially against a buffer containing 25 mM HEPES (pH 7.4), 100 mM NaCl, and 10 mM EDTA, then against 20 mM HEPES (pH 7.4), 150 mM NaCl, and 50% (v/v) glycerol, and stored at −20°C. FIXa concentrations were determined by antithrombin titration in the presence of heparin as previously described.29 The concentration of FIXaEGR was determined using the Bradford method.30
Other proteins employed in this study
Recombinant B-domain–deleted FVIII was purified with the monoclonal antibody VK3431 and stored at −20°C in a buffer containing 20 mM HEPES (pH 7.4), 800 mM NaCl, 10 mM CaCl2 and 50% (v/v) glycerol. The A2 domain of FVIII was isolated from thrombin-activated FVIII by ion-exchange chromatography as previously described,10 with the exception that a Source S column was used instead of a Mono S column. FX and α-thrombin were obtained as previously described.32-34
Phospholipid vesicles
PS/PC vesicles were prepared by sonication as previously described.31 Vesicles contained 50% PS for kinetic studies and 15% PS for HDX-MS experiments.
FIXa catalytic activity
FIXa amidolytic activity was assessed using substrate CH3SO2-(D)-CHG-Gly-Arg-pNA in a buffer containing 33% (v/v) ethylene glycol, 100 mM NaCl, 10 mM CaCl2, 0.2% (w/v) HSA, 50 mM Tris (pH 7.4) at 37°C as previously described.27 FX activation was assessed in the presence of varying concentrations FVIIIa and FX in a buffer containing 100 mM NaCl, 10 mM CaCl2, 0.5% (w/v) ovalbumin, 50 mM Tris (pH 7.4) at 37°C as previously described.27 Data were fitted in the Michaelis-Menten equation to obtain Km and kcat values.
Surface plasmon resonance (SPR) analysis
SPR studies were performed using the BIAcore3000 Biosensor System and immobilized FVIII A2 domain. FIXa binding was assessed in a buffer containing 150 mM NaCl, 2 mM CaCl2, 0.005% (v/v) Tween-20, 20 mM HEPES (pH 7.4) at 25°C with a flow of 20 μl/min as described in detail elsewhere.27
HDX-MS
FIXa (1.7 μM) was incubated with FVIII (3.3 μM) and phospholipids (0.21 mM) in a buffer containing 20 mM HEPES (pH 7.4), 150 mM NaCl, 5 mM CaCl2 for 90 seconds at room temperature. Thrombin prediluted in the same buffer was added to a final concentration of 0.33 μM to allow FVIII activation for 60 seconds at 24°C. In the absence of FVIII, the FVIII fraction was substituted with FVIII storage buffer, but thrombin was still added. Samples were handled without prior lipid removal by a LEAP PAL robot (LEAP Technologies, Morrisville, NC). Samples (3 μl) were diluted 10-fold in D2O buffer (20 mM imidazole [pH 7.3], 133 mM NaCl, 5 mM CaCl2 in 99.9% D2O), allowing HDX for 10, 20, 30, 45, 60, 100, 150, 500, and 1000 seconds at 24°C. Water reference samples were diluted in 20 mM imidazole (pH 7.3), 133 mM NaCl, 5 mM CaCl2 in H2O. HDX experiments involving FIXa mutants used fewer incubation time points, usually 10, 30, and 100 seconds, in order to avoid interference by FVIIIa instability (half-life ∼10 min35 ). In all cases, after incubation, 25 μl of the deuterated mix was quenched at 4°C by the addition of 25 μl of 1.4 M TCEP-HCl and 2 M urea, adjusted to pH 2.5 using NaOH. Proteolytic digestion was performed inline using a Poroszyme Immobilized Pepsin Cartridge (Thermo Fisher Scientific) with an isocratic flow of 5% (v/v) acetonitrile, 0.1% (v/v) formic acid at 4°C. Peptides were trapped on an Acclaim Guard Column 120 C18, 5 µm, 2 × 10 mm (Thermo Fisher Scientific) and washed for 30 seconds. The trap was then switched with a 10-cm Hypersil GOLD C18 analytic column (Thermo Fisher Scientific) with a gradient from 10% to 80% of buffer B (0.1% [v/v] formic acid in 80% [v/v] acetonitrile). Eluted peptides were sprayed into an LTQ Orbitrap-XL (Thermo Fisher Scientific) mass spectrometer operating in the positive ion mode. Collision-induced dissociation was used to fragment the 3 most intense precursor ions with tandem mass spectrometry fragmentation.
Data analysis of HDX-MS
Sequence and retention times of nondeuterated peptides were analyzed using PEAKS (PEAKS 7.0; Bioinformatics Solutions Inc). Deuterated peptides were analyzed in HDExaminer 2.2.0 (Sierra Analytics). No back-exchange correction was performed because only the relative levels of deuterium incorporation of individual peptides were compared under various conditions. Obtained HDX data are the result of 3 to 6 independent measurements; the mean value was used and the standard deviation is displayed for each time point. In addition, heat maps were constructed to visualize HDX data (see the supplemental Data, available on the Blood Web site). The software Pymol (Schrödinger, Cambridge, MA) was used to map the peptides on a 3-dimensional model structure. To define the extent of a change we subdivided the obtained peptides into 3 categories: prominent change, moderate change, and no appreciable change (criteria are detailed in the figure legends).
Results
HDX of FIXa–FVIIIa complex
To allow the assembly of the FIXa–FVIIIa complex, FIXa was incubated with an excess of FVIII in the presence of thrombin, calcium ions, and phospholipids as indicated in “Materials and methods.” Our HDX-MS protocol compromised between the lower limit for protein concentrations for optimal recovery and identification of peptides and the upper limit for phospholipid concentration and PS content to still allow proper peptide separation prior to MS measurements. The experimental conditions allow an appreciable amount of complex formation, although in approximately fivefold excess of protein accumulated over FVIII-binding sites on the lipid membrane.36 We obtained 75 unique FIXa peptides (Figure 1) spanning from the EGF-2 domain throughout the protease domain. The coverage of the light chain and the catalytic domain was 33.1% and 98.7%, respectively (Figure 1). Because of the low intensity of Gla domain and the EGF-1 domain peptides of the light chain, fragments belonging to these regions were not consistently detectable and, therefore, were not suitable for further analysis. This is mainly because of the posttranslational modifications in the Gla domain (abundant negative charges because of γ-carboxylation) and the EGF-1 domain (glycosylation). Multiple replicates (3-6) showed that our data were highly reproducible. As for the EGF-2 and protease domain of FIXa, our data revealed multiple regions of the FIXa molecule that were affected by FVIIIa, covering a major part of the protease domain and EGF-2 (Figure 2). We categorized the extent of change in deuterium uptake as prominent, moderate, or no appreciable change. Eight peptides showed a prominent change (dark red in Figures 1 and 2), localizing in the 99-loopCT, 162-helixCT, and helix 236-240CT. Thirteen peptides showed a moderate change (yellow in Figures 1 and 2). These were located in the EGF-2 domain, at the interface of EGF2/protease domain, and helix 126-132CT, 148-loopCT, 170-loopCT, and 186-loopCT elsewhere in the protease domain. Finally, 54 peptides displayed no appreciable deuterium uptake difference in the presence of FVIIIa (light gray in Figure 1; see also supplemental Figure 1 for the full set of peptides). All observed FVIIIa-induced changes correspond to reduced deuterium uptake, which may be to either direct binding of FVIIIa or to secondary effects because of FVIIIa binding to another region in FIXa.

Coverage of HDX-MS of FIXa–FVIIIa complex. HDX-MS experiments involved 9 time points, and changes in deuterium uptake were rated as prominent when all 9 time points displayed a reduction in deuterium uptake per peptide of ≥0.35 Da for the entire peptide. These were mapped on FIXa sequence in red. Changes were rated moderate when 4 to 8 time points showed reduction by ≥0.35 Da were mapped in yellow. No appreciable change was designated by peptides with less than 3 time points with ≥0.35 Da difference in deuterium uptake. FIXa numbering is assigned underneath the sequence, with chymotrypsin numbering between brackets. FIXa domains and regions are also indicated. Gla residues are indicated in black bold characters. Glycosylation sites are shown in red.
Coverage of HDX-MS of FIXa–FVIIIa complex. HDX-MS experiments involved 9 time points, and changes in deuterium uptake were rated as prominent when all 9 time points displayed a reduction in deuterium uptake per peptide of ≥0.35 Da for the entire peptide. These were mapped on FIXa sequence in red. Changes were rated moderate when 4 to 8 time points showed reduction by ≥0.35 Da were mapped in yellow. No appreciable change was designated by peptides with less than 3 time points with ≥0.35 Da difference in deuterium uptake. FIXa numbering is assigned underneath the sequence, with chymotrypsin numbering between brackets. FIXa domains and regions are also indicated. Gla residues are indicated in black bold characters. Glycosylation sites are shown in red.
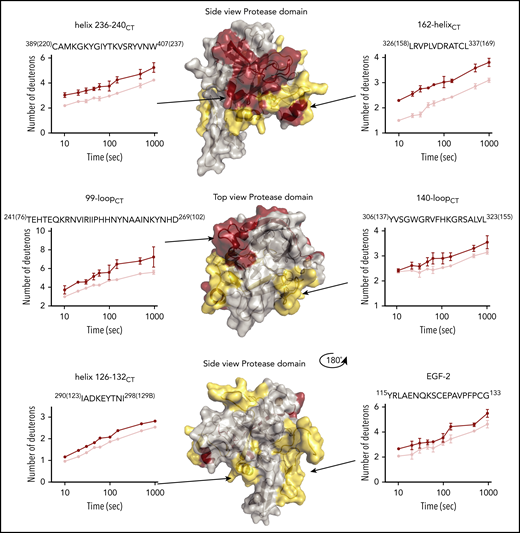
Deuterium uptake plots of HDX-MS of FIXa–FVIIIa complex. Changes in deuterium uptake in HDX-MS of FIXa–FVIIIa complex are displayed dark red (predominant change), yellow (moderate change), and gray on the crystal structure of FIXa, Protein Data Base (PDB) code 2wpm.14 Data indicate that major part of the protease domain and EGF-2 is affected by FVIIIa. Examples of deuterium uptake plots are also shown. Chymotrypsin numbering is indicated between brackets at the side of the peptide sequence. The red curves indicate FIXa in the absence of FVIIIa. FIXa incubated in the presence of FVIIIa is shown in pink. The complete set of peptides is reported in supplemental Figure 1.
Deuterium uptake plots of HDX-MS of FIXa–FVIIIa complex. Changes in deuterium uptake in HDX-MS of FIXa–FVIIIa complex are displayed dark red (predominant change), yellow (moderate change), and gray on the crystal structure of FIXa, Protein Data Base (PDB) code 2wpm.14 Data indicate that major part of the protease domain and EGF-2 is affected by FVIIIa. Examples of deuterium uptake plots are also shown. Chymotrypsin numbering is indicated between brackets at the side of the peptide sequence. The red curves indicate FIXa in the absence of FVIIIa. FIXa incubated in the presence of FVIIIa is shown in pink. The complete set of peptides is reported in supplemental Figure 1.
Similar experiments were performed in the absence of phospholipids. As shown in supplemental Figure 2, fewer changes were observed. Predominant changes were still observed in the 99-loopCT of the protease domain and in peptide F98-S110 of the EGF-2 domain, suggesting that FVIIIa and FIXa do assemble in solution. However, the absence of changes in other FIXa regions demonstrates that most of the changes detected in Figure 1 reflect the lipid-bound fraction of FIXa–FVIIIa complex.
HDX of active site-inhibited FIXa (FIXaEGR)
Changes in HDX were visualized in Figure 2 using 1 of the several available crystal structures of the EGF-2/protease domain segment, which all are based on active-site–inhibited FIXa frozen in a substrate-bound conformation.14 Similarly, we employed HDX to explore changes in FIXa occurring upon irreversible inhibition by the pseudo-substrate EGRck (FIXaEGR). Comparison of FIXaEGR with FIXa displayed a variety of changes, comprising both deuterium uptake reduction and increase. An increase in deuterium uptake for FIXaEGR was observed in the EGF-1/EGF-2 domain hinge (moderate increase; green in Figure 3A) and in the EGF-2/protease domain interface and 99-loopCT (prominent increase; blue in Figure 3A), whereas a decrease occurred in the 148-loopCT and 220-loopCT of the FIXaEGR (prominent; red in Figure 3A) and 162-helixCT regions (moderate; yellow in Figure 3A). Individual time courses are given in supplemental Figure 3.
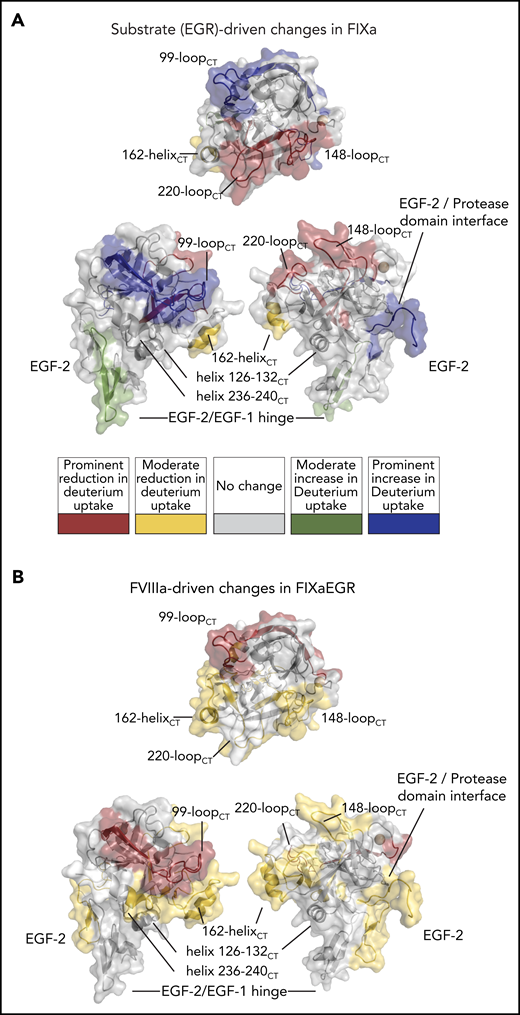
HDX-MS of FIXa irreversibly bound to the pseudo-substrate Glu-Gly-Arg-chloromethylketone (FIXaEGR). (A) FIXaEGR was compared with EGR-free FIXa where changes (mapped on FIXa crystal structure PDB: 2wpm) were detectable for hinge EGF-1/EGF-2, interface EGF-2/protease domain, 99-loopCT, 148-loopCT, 162-helixCT, and 220-loopCT. (B) FIXaEGR was incubated in the presence and absence of FVIIIa, and differences were highlighted on the FIXa crystal structure. Changes were detectable at EGF-2, interface EGF-2/protease domain, 99-loopCT, helix 126-132CT, 148-loopCT, 162-helixCT, and helix 236-240CT. HDX-MS measurements were based on 3 time points and mapped following the color coding as indicated. Differences were rated as prominent when 3 out of 3 time points showed ≥0.35 Da difference per peptide, moderate with 1 or 2 time points out of 3, and no appreciable change when all time points showed <0.35 Da difference.
HDX-MS of FIXa irreversibly bound to the pseudo-substrate Glu-Gly-Arg-chloromethylketone (FIXaEGR). (A) FIXaEGR was compared with EGR-free FIXa where changes (mapped on FIXa crystal structure PDB: 2wpm) were detectable for hinge EGF-1/EGF-2, interface EGF-2/protease domain, 99-loopCT, 148-loopCT, 162-helixCT, and 220-loopCT. (B) FIXaEGR was incubated in the presence and absence of FVIIIa, and differences were highlighted on the FIXa crystal structure. Changes were detectable at EGF-2, interface EGF-2/protease domain, 99-loopCT, helix 126-132CT, 148-loopCT, 162-helixCT, and helix 236-240CT. HDX-MS measurements were based on 3 time points and mapped following the color coding as indicated. Differences were rated as prominent when 3 out of 3 time points showed ≥0.35 Da difference per peptide, moderate with 1 or 2 time points out of 3, and no appreciable change when all time points showed <0.35 Da difference.
We further examined the effect of assembly of FIXaEGR with FVIIIa and lipids under the conditions of Figure 2. Differences between FIXaEGR in the presence and absence of FVIIIa proved to affect peptides very similarly to those in noninhibited FIXa in the presence of FVIIIa (Figure 3B vs Figure 2). The effect of assembly of FIXaEGR with FVIIIa is reflected by a prominent reduction of deuterium uptake at the 99-loopCT, and moderate reduction at the EGF-2 domain and the EGF-2/protease domain interface, and 148-loopCT, helix 126-132CT, 162-helixCT and 170-loopCT, 186-loopCT, and helix 236-240CT (supplemental Figures 3 and 4A-D).
Interestingly, the effects of EGR and FVIIIa are nonoverlapping in some FIXa regions. For instance, changes at 220-loopCT and EGF-1/EGF-2 hinge peptide F98-G114 are observed mainly upon EGR incorporation (Figure 4A). In contrast, reduced deuterium incorporation at EGF-2 peptide Y115-G133, helix 126-132CT, 162-helixCT, the 186-loopCT, and helix 236-240CT seem to be predominantly driven by FVIIIa (Figure 4B). Helices 126-132CT and 236-240CT are located close to the 162-helixCT, which has been previously implicated in FVIIIa binding.12,13 These 3 helices together form the basic surface patch also known as exosite II, which has been reported to bind heparin and possibly also FVIIIa.37
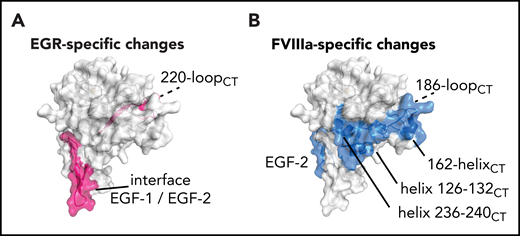
Cofactor-induced vs substrate-induced changes in deuterium uptake. (A) Among the changes induced by the EGRck alone, the 220-loopCT appears to be the only region involved on the protease domain together with the hinge EGF-1/EGF-2 on the light chain (pink). (B) FVIIIa induces changes on EGF-2, helix 126-132CT, helix 236-240CT, and the C-terminal part of 162-helixCT (blue). Data indicate the cluster of helices, helix 126-132CT, helix 236-240CT, and the C-terminal part of 162-helixCT, known as exosite II, as the putative FVIIIa binding site.
Cofactor-induced vs substrate-induced changes in deuterium uptake. (A) Among the changes induced by the EGRck alone, the 220-loopCT appears to be the only region involved on the protease domain together with the hinge EGF-1/EGF-2 on the light chain (pink). (B) FVIIIa induces changes on EGF-2, helix 126-132CT, helix 236-240CT, and the C-terminal part of 162-helixCT (blue). Data indicate the cluster of helices, helix 126-132CT, helix 236-240CT, and the C-terminal part of 162-helixCT, known as exosite II, as the putative FVIIIa binding site.
Functional properties of FIXa variants with substitutions in exosite II
To verify the involvement of the FIXa helix 126-132CT, 162-helixCT and 170-loopCT, and helix 236-240CT in FVIIIa-dependent catalysis, we selected the basic residues K293{126CT}, R333{165CT}, R338{170CT}, K341{173CT}, K400{230CT}, and R403{233CT} (Figure 5D) for substitution into alanine. These substitutions did not affect the activity toward the substrate CH3SO2-(D)-CHG-Gly-Arg-pNA (Table 1). The FIXa variants further displayed similar apparent Km values of FX activation in the presence of Ca2+ and phospholipids (Table 1), whereas kcat values were slightly reduced, in particular for FIXaR333A{165CT}. These data imply that these substitutions have limited impact on FIXa catalytic activity in the absence of FVIIIa.
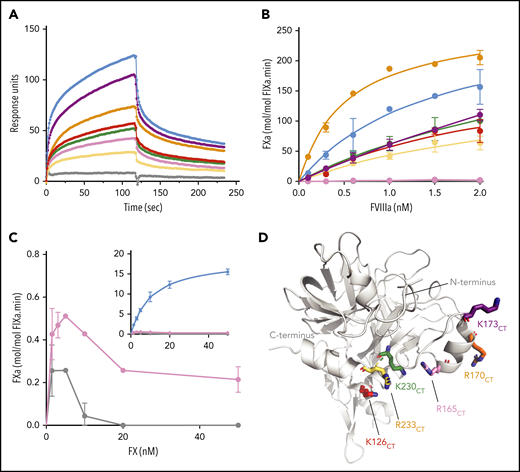
Functional characterization of FIXa with substitutions in exosite II. (A) Binding of FIXa variants to the A2 subunit of FVIII was measured by SPR as described in “Materials and methods.” The graph shows association and dissociation curves for wt-FIXa (blue), FIXaK293A{126CT} (red), FIXaR333A{165CT} (pink), FIXaR338A{170CT} (orange), FIXaK341A{173CT} (purple), FIXaK400A{230CT} (green), FIXaR403A{233CT} (yellow), and the double mutant FIXaR333A+R403A{165+233CT} (gray) at a concentration of 400 nM. (B) FX activation in the presence of FVIIIa was performed with various concentrations of FVIII. FVIII was incubated with phospholipids (100 µM), FX (200 nM), and 0.1 nM of wt-FIXa (blue), FIXaK293A{126CT} (red), FIXaR333A{165CT} (pink), FIXaR338A{170CT} (orange), FIXaK341A{173CT} (purple), FIXaK400A{230CT} (green), FIXaR403A{233CT} (yellow), or FIXaR333A+R403A{165+233CT} (gray). Data represent the mean of 2 independent experiments. (C) Activation of FX (0-50 nM) by 0.3 nM of FIXaR333A{165CT} (pink) or FIXaR333A+R403A{165+233CT} (gray). The inset shows the same data with wt-FIXa included. FX activation was assessed in the presence of 100 μM phospholipids and 0.3 nM FVIIIa. Data represent the mean of 2 to 3 independent experiments. (D) Basic residues of the α-helix 126-132CT, 162-helixCT, and α-helix 236-240CT were mutated into alanine residues. Chymotrypsin numbering is indicated (FIXa PDB 2wpm).
Functional characterization of FIXa with substitutions in exosite II. (A) Binding of FIXa variants to the A2 subunit of FVIII was measured by SPR as described in “Materials and methods.” The graph shows association and dissociation curves for wt-FIXa (blue), FIXaK293A{126CT} (red), FIXaR333A{165CT} (pink), FIXaR338A{170CT} (orange), FIXaK341A{173CT} (purple), FIXaK400A{230CT} (green), FIXaR403A{233CT} (yellow), and the double mutant FIXaR333A+R403A{165+233CT} (gray) at a concentration of 400 nM. (B) FX activation in the presence of FVIIIa was performed with various concentrations of FVIII. FVIII was incubated with phospholipids (100 µM), FX (200 nM), and 0.1 nM of wt-FIXa (blue), FIXaK293A{126CT} (red), FIXaR333A{165CT} (pink), FIXaR338A{170CT} (orange), FIXaK341A{173CT} (purple), FIXaK400A{230CT} (green), FIXaR403A{233CT} (yellow), or FIXaR333A+R403A{165+233CT} (gray). Data represent the mean of 2 independent experiments. (C) Activation of FX (0-50 nM) by 0.3 nM of FIXaR333A{165CT} (pink) or FIXaR333A+R403A{165+233CT} (gray). The inset shows the same data with wt-FIXa included. FX activation was assessed in the presence of 100 μM phospholipids and 0.3 nM FVIIIa. Data represent the mean of 2 to 3 independent experiments. (D) Basic residues of the α-helix 126-132CT, 162-helixCT, and α-helix 236-240CT were mutated into alanine residues. Chymotrypsin numbering is indicated (FIXa PDB 2wpm).
Functional properties of FIXa variants with substitutions in exosite II
| FIXa variant | Amidolytic activity | FX activation in the absence of FVIIIa | FX activation in the presence of FVIIIa | |||
|---|---|---|---|---|---|---|
| Apparent Km (mM) | kcat (s−1) | Apparent Km (nM) | 103 × kcat (min−1) | Apparent Km (nM) | kcat (min−1) | |
| wt-FIXa | 2.1 ± 0.3 | 13.2 ± 0.5 | 91 ± 6 | 25 ± 1 | 19 ± 6 | 54 ± 4 |
| FIXaK126ACT | 2.4 ± 0.3 | 10.9 ± 1.7 | 83 ± 19 | 15 ± 1 | 4 ± 1 | 26 ± 1 |
| FIXaR165ACT | 3.6 ± 0.5 | 11.1 ± 0.8 | 55 ± 9 | 7 ± 1 | ND | ND |
| FIXaR170ACT | 1.7 ± 0.2 | 11 ± 0.8 | 68 ± 15 | 17 ± 1 | 20 ± 6 | 100 ± 7 |
| FIXaK173ACT | 1.9 ± 0.2 | 9.3 ± 0.6 | 82 ± 12 | 11 ± 1 | 9 ± 5 | 39 ± 2 |
| FIXaK230ACT | 3.9 ± 0.7 | 13.3 ± 2.8 | 53 ± 13 | 14 ± 1 | 9 ± 2 | 30 ± 2 |
| FIXaR233ACT | 2.8 ± 0.3 | 11.4 ± 1.3 | 90 ± 24 | 14 ± 1 | 3 ± 1 | 17 ± 1 |
| FIXaR165A+R233ACT | 9.6 ± 3.3 | 14.2 ± 0.8 | 61 ± 14 | 5 ± 1 | ND | ND |
| FIXa variant | Amidolytic activity | FX activation in the absence of FVIIIa | FX activation in the presence of FVIIIa | |||
|---|---|---|---|---|---|---|
| Apparent Km (mM) | kcat (s−1) | Apparent Km (nM) | 103 × kcat (min−1) | Apparent Km (nM) | kcat (min−1) | |
| wt-FIXa | 2.1 ± 0.3 | 13.2 ± 0.5 | 91 ± 6 | 25 ± 1 | 19 ± 6 | 54 ± 4 |
| FIXaK126ACT | 2.4 ± 0.3 | 10.9 ± 1.7 | 83 ± 19 | 15 ± 1 | 4 ± 1 | 26 ± 1 |
| FIXaR165ACT | 3.6 ± 0.5 | 11.1 ± 0.8 | 55 ± 9 | 7 ± 1 | ND | ND |
| FIXaR170ACT | 1.7 ± 0.2 | 11 ± 0.8 | 68 ± 15 | 17 ± 1 | 20 ± 6 | 100 ± 7 |
| FIXaK173ACT | 1.9 ± 0.2 | 9.3 ± 0.6 | 82 ± 12 | 11 ± 1 | 9 ± 5 | 39 ± 2 |
| FIXaK230ACT | 3.9 ± 0.7 | 13.3 ± 2.8 | 53 ± 13 | 14 ± 1 | 9 ± 2 | 30 ± 2 |
| FIXaR233ACT | 2.8 ± 0.3 | 11.4 ± 1.3 | 90 ± 24 | 14 ± 1 | 3 ± 1 | 17 ± 1 |
| FIXaR165A+R233ACT | 9.6 ± 3.3 | 14.2 ± 0.8 | 61 ± 14 | 5 ± 1 | ND | ND |
Catalytic efficiency of FIXa variants of the exosite II was evaluated toward varying concentrations of the peptide substrate CH3SO2-(D)-CHG-Gly-Arg-pNA (indicated as amidolytic activity) and of the natural substrate FX in the presence of phospholipids and Ca2+ ions in the absence and presence of FVIIIa. Experimental conditions are given in “Materials and methods” and in the legend to Figure 5. Data were fitted in the Michaelis-Menten equation to obtain Km and kcat values. Kinetic parameters represent the mean ± standard deviation of 2 to 3 independent experiments.
ND, not determined because of substrate inhibition; see Figure 5C.
Because the 162-helixCT was previously proposed to bind to the A2 domain,12 we assessed the contribution of the single substitutions of exosite II in FVIIIa A2 domain binding by SPR experiments. Individual association and dissociation curves displayed heterogeneous kinetics, and could serve for qualitative comparison only. Figure 5A shows that all single substitutions reduced the association with the immobilized A2 domain. However, none of the substitutions fully abolished A2 domain binding. The most pronounced A2 domain association defects were observed for FIXaR403A{233CT} and, to a lesser extent, for FIXaR333A{165CT}, whereas A2 domain binding was fully abolished by the double substitution FIXaR333A+R403A{165+233CT}. These data suggest that multiple residues within exosite II contribute to an extended FVIIIa binding interface.
FIXa mutants were further characterized to assess their FVIIIa-assisted FX activation. FVIIIa titration experiments showed dose-dependent acceleration of FX activation by wild-type FIXa (wild-type [wt]-FIXa) by several thousandfold (Figure 5B; Table 1). Four FIXa variants displayed reduced FX activation that seemed compatible with their reduced A2 domain association (Figure 5A). One variant, FIXaR338A{170CT}, combined slightly reduced A2 domain interaction with increased response to FVIIIa (Figure 5B) and twofold increase in kcat (Table 1). The most prominent exception was FIXaR333A{165CT}, which, despite its residual A2 domain association being substantially conserved, lacked any response to FVIIIa (Figure 5B). In FX titration experiments, 4 of the FIXa mutants displayed typical substrate-dependent rate increase, although kcat was lower than for wt-FIXa (Table 1). However, the 2 mutants comprising the R333A{165CT} substitution displayed abnormal kinetics in that increasing the FX concentration actually reduced activity (Figure 5C). This substrate inhibition suggests that FIXaR333A{165CT} displays a more general defect in assembling the FIXa–FVIIIa–FX complex. These data suggest that residue R333{165CT}, although contributing to FVIIIa A2 domain interaction, is particularly crucial for FVIIIa-driven rate enhancement of FIXa.
HDX of FVIIIa assembly with FIXaR333A{165CT}
We first addressed the possibility that the FIXaR333A{165CT} mutant displays general defects at the backbone level. In comparison with wt-FIXa, FIXaR333A{165CT} displayed a prominent reduction in deuterium incorporation in peptides at the interface between the EGF-1/EGF-2 domains and the EGF-2/protease domain (red in Figure 6A). Moderate reduction occurred in surface loops, in particular at 70-loopCT, 99-loopCT, 148-loopCT, 170-loopCT, and 186-loopCT (yellow in Figure 6A). We further compared FIXaR333A{165CT} in the presence and absence of FVIIIa. Interestingly, only few FVIIIa-driven changes occurred (Figure 6B-C). A minor reduction of deuterium uptake was observed for the 99-loopCT only, whereas FVIIIa-induced changes at helix 126-132CT, 162-helixCT, 170-loopCT, 186-loopCT, and helix 236-240CT that occur in wt-FIXa (Figure 2) were no longer apparent (Figure 6C; supplemental Figure 4E-F).
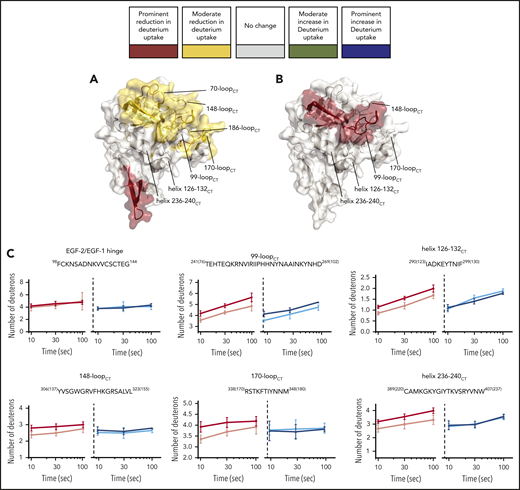
HDX-MS of the heavy-chain variant FIXaR333A{165CT}. FIXaR333A{165CT} ability to interact with FVIIIa in the FIXa–FVIIIa complex was investigated with HDX-MS. (A-B) FIXa protease domain and EGF-2 (PDB: 2wpm). The deuterium uptake changes are color coded according to their intensity as described in Figure 3. (A) Deuterium uptake differences in FIXaR333A{165CT} compared with wt-FIXa. (B) Deuterium uptake differences in FIXaR333A{165CT} compared with FIXaR333A{165CT} in the presence of FVIIIa. (C) Example plots comparing deuterium uptake differences between wt-FIXa, in the absence (red) or presence (pink) of FVIIIa, and FIXaR333A{165CT}, in the absence (dark blue) or presence (light blue) of FVIIIa.
HDX-MS of the heavy-chain variant FIXaR333A{165CT}. FIXaR333A{165CT} ability to interact with FVIIIa in the FIXa–FVIIIa complex was investigated with HDX-MS. (A-B) FIXa protease domain and EGF-2 (PDB: 2wpm). The deuterium uptake changes are color coded according to their intensity as described in Figure 3. (A) Deuterium uptake differences in FIXaR333A{165CT} compared with wt-FIXa. (B) Deuterium uptake differences in FIXaR333A{165CT} compared with FIXaR333A{165CT} in the presence of FVIIIa. (C) Example plots comparing deuterium uptake differences between wt-FIXa, in the absence (red) or presence (pink) of FVIIIa, and FIXaR333A{165CT}, in the absence (dark blue) or presence (light blue) of FVIIIa.
HDX of FVIIIa assembly with FIXaE78K
In addition to FIXaR333A{165CT}, other FIXa variants have been described with reduced response to FVIIIa.9,10 Interestingly, these comprise substitutions that are located beyond the FIXa protease domain, at the hinge between the EGF-1 and EGF-2 domains in the light chain.9 One of these, FIXaE78K, was included in this study. In comparison with wt-FIXa, FIXaE78K displayed a moderate increase in deuterium uptake at the bottom of the EGF-2 domain (green in Figure 7A). The variant further displayed a moderate deuterium incorporation increase at the 186-loopCT level. No other differences were observed for the FIXaE78K variant. When comparing the variant in the absence and presence of FVIIIa (Figure 7C; supplemental Figure 4G-H), FIXaE78K displayed a FVIIIa-driven reduction of deuterium uptake in helix 236-240CT and the 99-loopCT (Figure 7B), although less pronounced than in wt-FIXa. In contrast, the typical FVIIIa-driven changes at helix 126-132CT, 162-helixCT, and 170-loopCT were lacking.
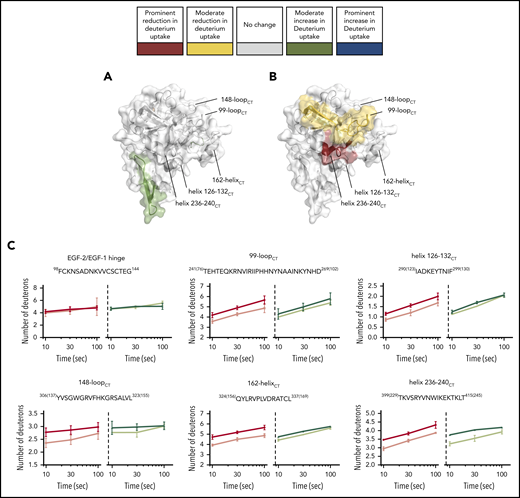
HDX-MS of the light-chain variant FIXaE78K. The ability of FIXaE78K to interact with FVIIIa in the FIXa–FVIIIa complex was investigated with HDX-MS. (A-B) Protease domain and EGF-2 are shown (PDB: 2wpm). The deuterium uptake changes are color coded according to their intensity as described in Figure 3. (A) Deuterium uptake differences in FIXaE78K compared with wt-FIXa. (B) Deuterium uptake differences in FIXaE78K compared with FIXaE78K in the presence of FVIIIa. (C) Example plots comparing deuterium uptake differences between wt-FIXa, in the absence (red) or presence (pink) of FVIIIa, and FIXaE78K, in the absence (dark green) or presence (light green) of FVIIIa.
HDX-MS of the light-chain variant FIXaE78K. The ability of FIXaE78K to interact with FVIIIa in the FIXa–FVIIIa complex was investigated with HDX-MS. (A-B) Protease domain and EGF-2 are shown (PDB: 2wpm). The deuterium uptake changes are color coded according to their intensity as described in Figure 3. (A) Deuterium uptake differences in FIXaE78K compared with wt-FIXa. (B) Deuterium uptake differences in FIXaE78K compared with FIXaE78K in the presence of FVIIIa. (C) Example plots comparing deuterium uptake differences between wt-FIXa, in the absence (red) or presence (pink) of FVIIIa, and FIXaE78K, in the absence (dark green) or presence (light green) of FVIIIa.
Discussion
Blood coagulation involves a cascade wherein activated coagulation proteases act in concert with their specific cofactor. Apart from the FIXa–FVIIIa complex, these include the FVIIa–tissue factor (TF) complex and the prothrombinase complex.2 Understanding the assembly of these enzyme–cofactor complexes and the mechanism of enhancement of enzyme function has remained a challenge for decades. In comparison with the FVIIa–TF complex, the FIXa–FVIIIa complex has remained poorly documented. The crystal structure of FVIIa complexed with a truncated TF has been resolved, which has greatly assisted the interpretation of later studies.38 The FVIIa–TF complex has also been studied by HDX-MS, and this has revealed an extensive allosteric network in FVIIa.26 For FIXa and FVIIIa, crystallography and modeling have established structures of the isolated constituents, but experimental data on the complex are limited so far.4,5,14,15,39,40
In this study, we employed HDX-MS to assess the implications of FVIIIa assembly with FIXa on lipid membranes. Of the numerous changes observed, some appeared predominantly FVIIIa-specific. These include helix 126-132CT, 162-helixCT, and helix 236-240CT in the basic exosite II, and probably reflect the footprint of the FVIIIa A2 domain on the catalytic domain (Figures 4 and 5). Other changes were driven by both FVIIIa and EGRck, and are considered to be allosteric. These comprise EGF-2 region Y115-G133, 99-loopCT, 148-loopCT, N-terminal part of the 162-helixCT, and 186-loopCT (Figure 3). This pattern is analogous to that in FVIIa in many respects. For instance, both FIXa and FVIIa display changes in the EGF-2 domain and 148-loopCT upon cofactor binding. Similarly, the allosteric effects of active site occupancy extend into the light chain in both FIXa and FVIIa. This interdomain crosstalk became apparent by increased deuterium exchange in FIXa (Figure 3A), whereas a decrease has been reported for FVIIa.26 Increased flexibility in the EGF-2 domain was also observed in the variant FIXaE78K (Figure 7). This seems not surprising, because this mutation disrupts the contact with R94 at the EGF-1/EGF-2 hinge.4,9 Remarkably, the compromised light chain in FIXaE78K also disrupts the binding on exosite II (Figure 7). It seems possible that wrenching the hinge between EGF-1 and EGF-2 causes silencing of the crosstalk between light chain and heavy chain. Alternatively, it may impair the FVIIIa-binding conformation to an extent that also impedes the subsequent docking of the A2 domain to exosite II. Because we did not recover peptides of the Gla-EGF1 segment, it remains difficult to distinguish between these possibilities.
A common activation-induced change concerns the 99-loopCT, which is prominent in FIXa, and has also been found in FVIIa and thrombin.17,26 In FIXa, this loop is believed to obstruct the access to the FIXa active site and needs to be unlocked for effective catalysis.5,41,42 Interestingly, in active-site–inhibited FIXa, deuterium uptake was increased, whereas the presence of FVIIIa reduced deuterium uptake (Figures 2 and 3B). This suggests that EGR binding brings this loop in an open, but flexible conformation, and that FVIIIa subsequently stabilizes this opened conformation, thus allowing free entrance of the substrate FX to the active site. As such, the cofactor-driven stabilization of this loop seems more dynamic in FIXa-FVIIIa than in the FVIIa–TF complex.26 This might be because the 99-loopCT in FIXa is longer than in its related coagulation enzymes.41,43
Our approach further revealed analogy with FVIIa in terms of binding interfaces on the protease domain. However, in comparison with the FVIIa-TF crystal structure,38 the binding region in FIXa is more extended, because also helix 236-240CT seems involved in the interaction with FVIIIa (Figures 4B and 5). Possibly, this is because of different cofactor dimensions, because TF is much smaller than FVIII.38 Nevertheless, the patterns of allosteric changes detected in HDX-MS in FVIIa and FIXa display striking similarity. In contrast to TF, activated factor V (FVa) in the prothrombinase complex does display homology with FVIII. Although the structure of the FVa–FXa complex has not been resolved, a crystal structure is available of a homologous enzyme–cofactor complex that occurs in snake Pseudonaja textilis.44 This displays a binding interface at exosite II that also includes helix 236-240CT of the FX homolog. This helix seems to be involved with an acidic surface in its FVa counterpart that is also present in FVIIIa. Interestingly, most of the residues at the exosite II region that we selected for site-directed mutagenesis are reported hemophilia B variants with defective activity.45 This phenotype is compatible with reduced FVIIIa A2 domain binding and FX activation (Figure 5).
One key modulator of FIXa activity proved to be residue R333{165CT}. Strikingly, FIXaR333A{165CT} not only revealed reduced FVIIIa binding (Figure 5A), but in contrast to other exosite II variants, the mutant also displayed prominent substrate inhibition (Figure 5C). This suggests unproductive FX binding to the compromised FIXa–FVIIIa complex and implies a dual role of 162-helixCT in assembly with both cofactor and substrate. It is noteworthy that the R338A{170CT} substitution, which is located in the same helix, is associated with gain-of-function in the presence of FVIIIa (Figure 5B; Table 1). This confirms an earlier study,46 and agrees with the notion that the substitutions R338L (FIX Padua) and R338Q (FIX Shanghai) display supranormal activity.47,48 Apparently, mutations in this helix may have opposite effects. Possibly, mutations of 162-helixCT alter the FVIIIa-driven allosteric signaling to the active site that influences substrate turnover. In this regard, it is interesting that the variant R333A{165CT} misses the typical FVIIIa-driven change in the 186-loopCT (Figure 6). Presumably, the disulphide bridge C168CT-C182CT accounts for the communication line between the 162-helixCT and the active site.15 In support of this view, overlapping peptides spanning the 162-helixCT identified the segment TCL167-169CT as mediating the prominent FVIIIa-dependent response (Figure 1). Similarly, the changes in the 186-loopCT largely depend on the residues FC181-182CT (supplemental Figure 1).
One surprising observation was that HDX-MS revealed a tangible consequence of the R333{165CT} mutation (Figure 6A). In comparison with wt-FIXa, the R333A{165CT} variant displayed a general reduction of deuterium exchange in multiple surface loops, suggesting the protease domain to be more rigid. It seems conceivable that, although this variant is not defective in amidolytic activity (Table 1), its overall rigidity contributes to the assembly defect with the natural substrate FX in the presence of FVIIIa. In this regard, it would be interesting to address the effect of FX on FIXa-FVIIIa assembly by HDX-MS. However, it proved challenging to achieve proper FIX peptide separation in the presence of membranes and an excess of FVIII-derived peptides; increasing complexity by including FX does not seem feasible at this stage. Our extensive sampling handling procedure prior to MS analysis has the inherent limitation that back-exchange significantly reduces the apparent extent of deuterium incorporation. Consequently, as in other studies employing a similar protocol,17,24,26,49 we report changes on the peptide level, whereas HDX-MS should have the potential of near-residue resolution. However, this will require more rigorous strategies to reduce back-exchange.50 Another issue is the lack of recovery of peptides from Gla-EGF-1 region. The post-translational modifications therein require complementary MS protocols to analyze membrane and FVIIIa binding to this part of the FIXa light chain. Despite these limitations, this study demonstrates that HDX-MS allows the assessment of structural consequences of single amino acid substitutions. As such, it may also prove useful for addressing the structural impact of disease-associated mutations in other hemostatic proteins.
For original data, please e-mail the corresponding author, Koen Mertens (k.mertens@sanquin.nl).
The online version of this article contains a data supplement.
The publication costs of this article were defrayed in part by page charge payment. Therefore, and solely to indicate this fact, this article is hereby marked “advertisement” in accordance with 18 USC section 1734.
Acknowledgment
This study was funded by the Utrecht Institute for Pharmaceutical Sciences, and by the Landsteiner Stichting voor Bloedtransfusie Research (LSBR 1417).
Authorship
Contribution: N.F., A.B.M., M.v.d.B., and K.M. designed the study; N.F., C.F., and M.B.-S. performed the experimental work; F.P.J.v.A., J.v.G., and E.H.T.M.E. developed the HDX-MS protocol; N.F., J.v.G., E.H.T.M.E., C.F., A.B.M., M.v.d.B., and K.M. analyzed and interpreted the data; N.F. made the figures; and N.F., A.B.M., M.v.d.B., and K.M. wrote the manuscript.
Conflict-of-interest disclosure: The authors declare no competing financial interests.
Correspondence: Koen Mertens, Department of Molecular and Cellular Hemostasis, Sanquin Research, Plesmanlaan 125, 1066 CX Amsterdam, The Netherlands; e-mail: k.mertens@sanquin.nl.

This feature is available to Subscribers Only
Sign In or Create an Account Close Modal