

TO THE EDITOR:
The mechanism of hemolysis in glucose 6-phosphate dehydrogenase (G6PD) deficiency is oxidative damage; in paroxysmal nocturnal hemoglobinuria (PNH), instead, red blood cells (RBCs) lyse when attacked by activated complement. The exquisite susceptibility of PNH RBCs to activated complement,1,2 resulting from lack of the complement regulators CD55 and CD59 on PNH cells, is the main mechanism of intravascular hemolysis and anemia in PNH.3,4 Intravascular hemolysis and its clinical consequences are effectively controlled by monoclonal antibodies (eg, eculizumab, ravulizumab) targeting complement C5.5-7 G6PD-deficient RBCs, being hypersusceptible to oxidative damage, undergo acute hemolysis when an exogenous trigger is applied.8,9 It has been suggested that oxidative damage could play a role in PNH as well,10,11 although it is susceptibility to complement-mediated lysis that dominates the picture. PNH is a very rare disease; however, G6PD deficiency is a relatively common X-linked genetic abnormality that in some countries may affect up to 30% of the population.9
We were prompted to investigate the interaction between complement activation and oxidative damage by a patient with PNH who was also G6PD-deficient. This patient, a 40-year-old woman from Sardinia, Italy, presented with increasing thrombocytopenia (platelets dropping from 100 to 40 × 109/L), mild granulocytopenia, and anemia; initially, the patient had no signs of hemolysis, and a flow cytometry analysis from another laboratory did not report any glycosylphosphatidylinositol-negative (PNH) cells. Two years later, she developed florid hemolytic PNH, with falling hemoglobin levels, lactate dehydrogenase up to 5 × upper limit of normal, high reticulocytosis, and 59% PNH granulocytes. Over the following 6 months, the PNH granulocyte population increased to 95%; gradual expansion of the PNH populations was associated with reversal of thrombocytopenia and neutropenia but with the payload of increasing intravascular hemolysis. The course of this patient illustrates well the evolution from aplastic anemia to PNH, probably the rule rather than the exception in the natural history of PNH.12 Six months after the PNH diagnosis, the patient needed RBC transfusions, and eculizumab was started at standard dosage. Lactate dehydrogenase levels promptly returned to normal, but reticulocytes did not (steadily above 250 × 109/L), and over 6 years, the packed RBC transfusion requirement was ∼10 units per year. PNH RBCs rose steadily from 20% (before eculizumab) to 86% on eculizumab. In addition, as in almost all patients on eculizumab,13 PNH RBCs became bound with complement C3 fragments, and their fraction rose to 57%. Thus, the patient had a minor response to eculizumab14 despite no evidence of bone marrow failure.
A peripheral blood smear (in steady state on eculizumab) revealed RBC morphology (Figure 1A) reminiscent of oxidative damage as seen in G6PD-deficient patients during a hemolytic attack. RBC G6PD activity was 50% of normal (5 IU/g hemoglobin). From DNA analysis, the patient was heterozygous for G6PD Mediterranean (G6PD-Med),15 the most common variant in Sardinia (Figure 1B). Women heterozygous for a mutant G6PD are epigenetic mosaics, as they have cells that express either the wild-type or the mutant G6PD transcript. PNH cells derive from a single hematopoietic stem cell with a somatic mutation in the X-linked PIGA gene16 ; thus, the clonal progeny of that cell is expected to express a single type of G6PD transcript. Indeed, restriction enzyme analysis of the G6PD complementary DNA from the PNH granulocytes and monocytes exhibited only the G6PD-Med transcript (Figure 1C). This indicates that the patient’s PIGA mutation (a frameshift 930T deletion in exon 4 causing a stop at aa329) must have occurred in a hematopoietic stem cell in which the active X chromosome carried the G6PD-Med allele. Because almost all granulocytes/monocytes and a very large fraction of RBCs in this patient were PNH, most of her blood cells were G6PD-deficient, just like in a hemizygous G6PD-Med male subject. Thus, in this patient, a large proportion of the RBCs has both the PNH abnormality and the G6PD deficiency; we hypothesized that her poor response to eculizumab might result from this unique interaction.
![Hematologic and molecular studies. (A) Peripheral blood smear (May-Grünwald-Giemsa). At low magnification (×100), anisocytosis and spherocytes are conspicuous (i). At high magnification (×1000), one sees spherocytes (ii), macrocytes (iii), hemighost (iv), and a nucleated red cell (v). (B) Restriction analysis of genomic DNA for G6PD-Med variant (Exon 6; 563 C>T; 188Ser>Phe).25 After MboII digestion of an amplicon including exons 6, intron 6, and exon 7, the restriction fragments expected from wild-type (WT) G6PD (377 bp) and from mutant G6PD-Med allele (277 bp) are indicated. Controls (WT, heterozygote, and hemizygote) and patient samples are shown; the patient is heterozygous for G6PD-Med. MW, molecular-weight size marker 100 bp DNA ladder; the brighter band is 500 bp. (C) Complementary DNA restriction analysis of G6PD transcripts. Granulocytes (polymorphonuclear neutrophils [PMN]) were isolated by using the double-density centrifugation method; monocytes (Mø) were obtained by using an adherence technique from peripheral blood mononuclear cells. At the time of sampling, 99% of granulocytes and 97% of monocytes were PNH. Normal (non-PNH) lymphocytes (L) were obtained after monocyte-depletion of peripheral blood mononuclear cells. G6PD complementary DNA obtained from the selected blood cell populations was amplified, and an amplicon comprising exons 4 to 7 was digested with MboII. The restriction fragments relevant for the identification of normal (246 bp) and mutant G6PD-Med (147 bp) transcripts are indicated (the 208 bp fragment arises from either transcripts). The patient’s PNH granulocytes and monocytes expressed only the G6PD-Med transcript. The patient’s normal (non-PNH) lymphocytes also expressed predominantely the G6PD-Med transcript, suggesting that in this patient, X inactivation in hematopoietic cells had been highly skewed toward G6PD deficiency, regardless of PNH. MW, molecular-weight size marker 100 bp DNA ladder; the upper band is 500 bp.](https://ash.silverchair-cdn.com/ash/content_public/journal/blood/136/26/10.1182_blood.2020007780/1/m_bloodbld2020007780f1.png?Expires=1767920058&Signature=Qyf~caiRIEudty899CiVQYgA8T4iBiYdioqxGGpEdJDBJmhKSZQZH6yzodEQjCNpH4xu8FPERJqQocggzCvpfuPFERA0pFnGEX7i47VCgs9SAfy--Plb9pRmyfHxN4iH8dGctpAYfHRzgUhsn5Sft~BOc77NoVFp7gYd8ZyStZ6cFvZsHPwPoQzE37lkg8bSloem2Etjl2mKnhY9GCQPKwCgn6lSKEL7rmVONh0qcOIerhQXBhs84Au3CORmai0fjHEwjNnpOKHZGWFtdDq9Ayu3yjrjMu-gXX7VWj2c74XldDY2rJmnGht8N8~8gJOVHvsylfqJezBWrIqLbvHbNQ__&Key-Pair-Id=APKAIE5G5CRDK6RD3PGA)
Hematologic and molecular studies. (A) Peripheral blood smear (May-Grünwald-Giemsa). At low magnification (×100), anisocytosis and spherocytes are conspicuous (i). At high magnification (×1000), one sees spherocytes (ii), macrocytes (iii), hemighost (iv), and a nucleated red cell (v). (B) Restriction analysis of genomic DNA for G6PD-Med variant (Exon 6; 563 C>T; 188Ser>Phe).25 After MboII digestion of an amplicon including exons 6, intron 6, and exon 7, the restriction fragments expected from wild-type (WT) G6PD (377 bp) and from mutant G6PD-Med allele (277 bp) are indicated. Controls (WT, heterozygote, and hemizygote) and patient samples are shown; the patient is heterozygous for G6PD-Med. MW, molecular-weight size marker 100 bp DNA ladder; the brighter band is 500 bp. (C) Complementary DNA restriction analysis of G6PD transcripts. Granulocytes (polymorphonuclear neutrophils [PMN]) were isolated by using the double-density centrifugation method; monocytes (Mø) were obtained by using an adherence technique from peripheral blood mononuclear cells. At the time of sampling, 99% of granulocytes and 97% of monocytes were PNH. Normal (non-PNH) lymphocytes (L) were obtained after monocyte-depletion of peripheral blood mononuclear cells. G6PD complementary DNA obtained from the selected blood cell populations was amplified, and an amplicon comprising exons 4 to 7 was digested with MboII. The restriction fragments relevant for the identification of normal (246 bp) and mutant G6PD-Med (147 bp) transcripts are indicated (the 208 bp fragment arises from either transcripts). The patient’s PNH granulocytes and monocytes expressed only the G6PD-Med transcript. The patient’s normal (non-PNH) lymphocytes also expressed predominantely the G6PD-Med transcript, suggesting that in this patient, X inactivation in hematopoietic cells had been highly skewed toward G6PD deficiency, regardless of PNH. MW, molecular-weight size marker 100 bp DNA ladder; the upper band is 500 bp.
Hematologic and molecular studies. (A) Peripheral blood smear (May-Grünwald-Giemsa). At low magnification (×100), anisocytosis and spherocytes are conspicuous (i). At high magnification (×1000), one sees spherocytes (ii), macrocytes (iii), hemighost (iv), and a nucleated red cell (v). (B) Restriction analysis of genomic DNA for G6PD-Med variant (Exon 6; 563 C>T; 188Ser>Phe).25 After MboII digestion of an amplicon including exons 6, intron 6, and exon 7, the restriction fragments expected from wild-type (WT) G6PD (377 bp) and from mutant G6PD-Med allele (277 bp) are indicated. Controls (WT, heterozygote, and hemizygote) and patient samples are shown; the patient is heterozygous for G6PD-Med. MW, molecular-weight size marker 100 bp DNA ladder; the brighter band is 500 bp. (C) Complementary DNA restriction analysis of G6PD transcripts. Granulocytes (polymorphonuclear neutrophils [PMN]) were isolated by using the double-density centrifugation method; monocytes (Mø) were obtained by using an adherence technique from peripheral blood mononuclear cells. At the time of sampling, 99% of granulocytes and 97% of monocytes were PNH. Normal (non-PNH) lymphocytes (L) were obtained after monocyte-depletion of peripheral blood mononuclear cells. G6PD complementary DNA obtained from the selected blood cell populations was amplified, and an amplicon comprising exons 4 to 7 was digested with MboII. The restriction fragments relevant for the identification of normal (246 bp) and mutant G6PD-Med (147 bp) transcripts are indicated (the 208 bp fragment arises from either transcripts). The patient’s PNH granulocytes and monocytes expressed only the G6PD-Med transcript. The patient’s normal (non-PNH) lymphocytes also expressed predominantely the G6PD-Med transcript, suggesting that in this patient, X inactivation in hematopoietic cells had been highly skewed toward G6PD deficiency, regardless of PNH. MW, molecular-weight size marker 100 bp DNA ladder; the upper band is 500 bp.
To test this hypothesis, we measured reactive oxygen species (ROS) in normal RBCs and in RBCs made PNH-like by blocking CD55 and CD59 with specific antibodies.17 RBCs were collected from 8 G6PD-deficient male subjects (G6PD-Med hemizygotes) and 11 healthy control subjects after signed informed consent was provided (according to an institutional review board–approved protocol). Two percent RBC suspensions were prepared in pooled ABO-compatible sera with or without eculizumab (400 μg/mL); complement was activated by mild acidification.18
First, when RBCs were exposed to sera in which complement had been heat inactivated, ROS levels were similar in non-PNH (hereinafter “non-PNH” indicates RBCs not treated with anti-CD55 and -CD59 antibodies) and in PNH-like RBCs from control subjects and from G6PD-deficient male subjects (Figure 2A). ROS levels did not increase when non-PNH RBCs were exposed to activated complement, whereas, in the same condtions, all PNH-like RBCs were lysed. As expected, ROS levels were higher in G6PD-deficient RBCs; however, even in these cells, complement activation per se produced no increase in ROS levels. We next assayed ROS accumulated over 24 hours in complement-activated sera in which C5 was blocked by eculizumab. With G6PD-normal RBCs from healthy control subjects, there was no difference in ROS levels, regardless of whether the RBCs were non-PNH or PNH-like (Figure 2B). On the other hand, in similar conditions, ROS levels were significantly higher in PNH-like RBCs compared with non-PNH RBCs from G6PD-deficient male subjects (P < .008; Figure 2B). Thus, complement activation on the surface of PNH-like RBCs results in high levels of ROS when RBCs, being G6PD-deficient, are defective in antioxidant defense; this excess of ROS can only be seen when C5 is blocked, because otherwise the PNH RBCs would be lysed by activated complement.
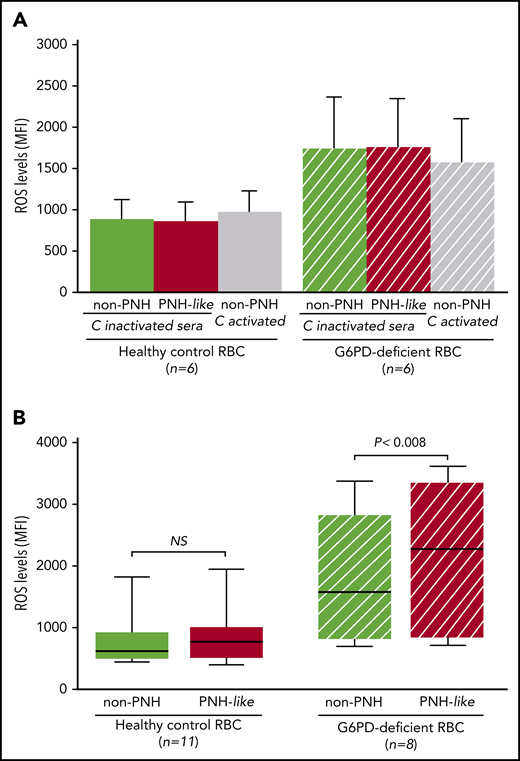
The effect of complement activation on oxidative stress in PNH-like RBCs, in the presence of C5 blockade, depends on their G6PD status. Levels of ROS, expressed as mean fluorescence intensity (MFI), were quantitated by flow cytometry with dichlorodihydrofluorescein diacetate (400 µM),10 after 24 hours at 37°C since complement activation. (A) Bar diagram of ROS levels in non-PNH (green bars) and in PNH-like RBCs (red bars) from 6 healthy control subjects (Healthy control RBC; solid bars) and from 6 G6PD-deficient male subjects (G6PD-deficient RBC; striped bars) after exposure to sera in which complement (C) had been heat-inactivated (C-inactivated sera). The gray bars show ROS levels when non-PNH RBCs were exposed to complement activated by acidification (C activated); as expected, PNH-like RBCs were lysed when exposed to activated complement (data not shown). Average and standard errors are shown. In these experiments, sera were acidified even when complement was due to be heat inactivated to make sure that acidification as such had no effect. (B) Box and whisker plot of levels of ROS after complement activation in the presence of C5 blockade by eculizumab in non-PNH (green boxes) and PNH-like RBCs (red boxes) from 11 healthy control subjects (Healthy control RBC; solid boxes) and from 8 G6PD-deficient male subjects (G6PD-deficient RBC; striped boxes). ROS levels are expressed as MFI, the bottom and top of the box show the 25th and 75th percentile, the horizontal line within the box shows the median, and the ends of the whiskers represent the minimum and the maximum value. Nonparametric tests for paired (Wilcoxon) and unpaired (Mann-Whitney) samples were performed, as appropriate. Statistical significance was accepted for P ≤ .05. NS, not statistically significant.
The effect of complement activation on oxidative stress in PNH-like RBCs, in the presence of C5 blockade, depends on their G6PD status. Levels of ROS, expressed as mean fluorescence intensity (MFI), were quantitated by flow cytometry with dichlorodihydrofluorescein diacetate (400 µM),10 after 24 hours at 37°C since complement activation. (A) Bar diagram of ROS levels in non-PNH (green bars) and in PNH-like RBCs (red bars) from 6 healthy control subjects (Healthy control RBC; solid bars) and from 6 G6PD-deficient male subjects (G6PD-deficient RBC; striped bars) after exposure to sera in which complement (C) had been heat-inactivated (C-inactivated sera). The gray bars show ROS levels when non-PNH RBCs were exposed to complement activated by acidification (C activated); as expected, PNH-like RBCs were lysed when exposed to activated complement (data not shown). Average and standard errors are shown. In these experiments, sera were acidified even when complement was due to be heat inactivated to make sure that acidification as such had no effect. (B) Box and whisker plot of levels of ROS after complement activation in the presence of C5 blockade by eculizumab in non-PNH (green boxes) and PNH-like RBCs (red boxes) from 11 healthy control subjects (Healthy control RBC; solid boxes) and from 8 G6PD-deficient male subjects (G6PD-deficient RBC; striped boxes). ROS levels are expressed as MFI, the bottom and top of the box show the 25th and 75th percentile, the horizontal line within the box shows the median, and the ends of the whiskers represent the minimum and the maximum value. Nonparametric tests for paired (Wilcoxon) and unpaired (Mann-Whitney) samples were performed, as appropriate. Statistical significance was accepted for P ≤ .05. NS, not statistically significant.
These findings strongly support our hypothesis that the poor response to eculizumab in our patient results from a unique interaction, within the same RBC, between the acquired PNH abnormality and her inherited G6PD deficiency. The precise mechanism is not yet clear. However, with C5 blockade, complement activation causes binding of C3 fragments to the surface of PNH RBCs both in vivo13 and in vitro.18 Upon complement activation in the presence of eculizumab, PNH-like G6PD-deficient RBCs become C3 bound, just as G6PD-normal RBCs (supplemental Figure 1). Moreover, C3 fragments on granulocytes elicit ROS formation19,20 ; thus, it is not unlikely that C3 fragments elicit ROS formation also on RBCs.
This interaction was not previously observed in 2 other patients with PNH and G6PD deficiency for different reasons. In 1 patient,21 who was on eculizumab, the very large population of PNH cells expressed the wild-type G6PD allele; the other patient was not on eculizumab.22 This interaction, which we have observed in vivo and validated in vitro, is novel and has pharmacogenetic implications. G6PD deficiency as such has only a mild impact on the clinical expression of PNH, as complement activation causes hemolysis of PNH RBCs regardless of their G6PD status (supplemental Figure 2A). Paradoxically, when the intravascular hemolysis of PNH RBCs is prevented by C5 blockade, activation of the proximal complement pathway results in oxidative damage; G6PD-deficient PNH RBCs are unable to cope with this damage, with consequent extravascular hemolysis (supplemental Figure 2B). This may be relevant in parts of the world where G6PD deficiency is common,9 and G6PD-deficient patients with PNH may become candidates for agents, currently under development,14,23 that target inhibition of C3 rather than C5.
The genetic polymorphism of G6PD was key to understanding the clonal origin of PNH cells.24 Fifty years later, we find that G6PD deficiency may influence the pathophysiology of PNH.
The online version of this article contains a data supplement.
Acknowledgments
The authors thank the patient and her family for their active participation and their positive attitude throughout the study. They also thank their Clinic staff for their support. BRIC216 and BRIC229 antibodies were a kind gift of David J. Anstee (Bristol Institute for Transfusion Science, Bristol, United Kingdom). Last, but not least, the authors thank the “Associazione Italiana Emoglobinuria Parossistica Notturna.”
Authorship
Contribution: M.S. performed the experiments and participated in designing the research, analyzing data, and writing the paper; A.P. and C.N. helped perform the research; M.D.A. helped perform the research and in analyzed data; G.C. and G.L.N. provided clinical care to the patient; and L.L. and R.N. designed the research, analyzed data, participated in the clinical management of the patient, and wrote the paper; and all authors reviewed the paper and ratified the final version.
Conflict-of-interest disclosure: R.N. has received lecture fees from Alexion Pharmaceuticals and served as a member of the Investigator Board for BioCryst. The remaining authors declare no competing financial interests.
Correspondence: Rosario Notaro, Laboratory of Cancer Genetics and Gene Transfer, Core Research Laboratory–Istituto per lo Studio, la Prevenzione e la Rete Oncologica (ISPRO), viale Pieraccini 6, 50139 Florence, Italy; e-mail: r.notaro@ispro.toscana.it.
