

Key Points
Mk TGFβ1 couples the number of hematopoietic stem and progenitor cells to the need for RBCs.
Interfering with the Mk TGFβ1 axis sensitizes erythropoiesis to low-dose EPO and increases RBC output.

Abstract
Erythropoietin (EPO) provides the major survival signal to maturing erythroid precursors (EPs) and is essential for terminal erythropoiesis. Nonetheless, progenitor cells can irreversibly commit to an erythroid fate well before EPO acts, risking inefficiency if these progenitors are unneeded to maintain red blood cell (RBC) counts. We identified a new modular organization of erythropoiesis and, for the first time, demonstrate that the pre-EPO module is coupled to late EPO-dependent erythropoiesis by megakaryocyte (Mk) signals. Disrupting megakaryocytic transforming growth factor β1 (Tgfb1) disorganized hematopoiesis by expanding the pre-EPO pool of progenitor cells and consequently triggering significant apoptosis of EPO-dependent EPs. Similarly, pharmacologic blockade of TGFβ signaling in normal mice boosted the pre-EPO module, leading to apoptosis of EPO-sensitive EPs. Subsequent treatment with low-dose EPO triggered robust RBC production in both models. This work reveals modular regulation of erythropoiesis and offers a new strategy for overcoming chronic anemias.
Introduction
It is estimated that one-third of the world’s population suffers from anemia.1,2 Daily production of >200 billion erythrocytes is required to keep up with routine losses, so even minor disequilibrium between blood loss and erythrocyte production can lead to anemia.3 Although many transient anemias are easily treated, therapy for chronic anemias is limited. Red blood cell (RBC) transfusions and erythropoiesis-stimulating agents are not always effective yet are linked to significant expense, inconvenience, potential toxicity, and generally transient utility.4,5 Despite these limitations, development of new approaches to treat chronic anemia has been limited by our incomplete understanding of erythropoiesis.
A great deal of what we know about erythropoiesis relates to the terminal maturation of erythroid precursors (EPs) leading to hemoglobinization, enucleation, and release of immature RBCs into the circulation.6,7 In the setting of anemia, or otherwise reduced oxygen delivery, juxtaglomerular renal erythropoietin (EPO)-producing cells can increase EPO secretion many thousands-fold, thereby serving as a feedback signal reporting erythrocyte need to the marrow.8-13 Nonetheless, EPO acts during a very narrow window of erythropoiesis, well after progenitor commitment to an exclusively erythroid fate.6,8,9 It is not known whether these final steps of RBC maturation are coupled to the earlier stages of hematopoietic stem and progenitor cell (HSPC) differentiation, a process that begins almost 3 weeks earlier when a hematopoietic stem cell (HSC) starts its march toward committed RBC precursors via a series of branching cell-fate decisions.7,14,15 In this study, we investigated whether terminal erythropoiesis is linked to the availability of EPO-insensitive HSPCs.
Materials and methods
Animals
Transgenic C57BL/6 mice with Cre recombinase driven from the regulatory elements of Cxcl4 (platelet factor 4 [Pf4]), C57BL/6-Tg(Pf4-cre)Q3Rsko/J (Pf4-cre),16 were crossed with mice harboring a floxed transforming growth factor β1 (TGFβ1) locus17 (Tgfb1tm2.1Doe, TGFβ1FL/FL mice) to generate Pf4-Cre/TGFβ1FL/FL mice with selective deletion of TGFβ1 in megakaryocytes (Mks) and platelets (TGFβ1ΔMk/ΔMk mice). GFAP-CreERT2 (B6;Cg-Tg(GFAP-cre/ERT2)505Fmv/J) mice with the human glial fibrillary acidic protein (GFAP) promoter sequence directing expression of a Cre-estrogen receptor (Cre-ERT2) fusion protein were bred into the TGFβ1FL/FL strain to generate GFAP-CreERT2/TGFβ1FL/FL mice (TGFβ1iSchwn/iSchwn). Deletion of Tgfb1 in Schwann cells (TGFβ1ΔSchwn/ΔSchwn) was induced at 5 weeks of age via intraperitoneal injection of tamoxifen (80 mg/kg; Millipore/Sigma-Aldrich) daily × 5 days followed by 4 weeks of tamoxifen chow (Envigo). TGFβ1ΔSchwn/ΔSchwn mice were rested for 1 week prior to analysis. Selective Cre activity was confirmed by immunofluorescence and/or flow cytometry by crossing the Cre-deleter strains (PF4-Cre or GFAP-CreERT2) into the Cre-reporter strain Rosa26-FSF-ZsGreen (B6;Cg-Gt(ROSA)26Sortm6(CAG-ZsGreen1)Hze/J).
Animals were maintained in the Weill Cornell Medicine Animal Facility. All protocols were approved by the Weill Cornell Medicine Institutional Animal Care and Use Committee.
Immunophenotypic analysis of bone marrow HSPCs and HSCs by flow cytometry
Mice were euthanized by CO2 asphyxiation; bones (femur, tibia, plus or minus humeri) were dissected free of muscle and tendons and crushed in Dulbecco modified Eagle medium using a mortar and pestle. The resulting cell suspension was filtered through a 40-µm mesh and washed in phosphate-buffered saline (PBS) EDTA buffer (2 mM EDTA, 0.2% bovine serum albumin in PBS, pH 7.4). Spleens were isolated, and then minced before grinding through a 40-µm mesh to generate single-cell suspensions. Lineage-negative (Lin−) cells were isolated using immunomagnetic beads using a biotinylated lineage cell depletion cocktail (Miltenyi Biotec).
Lin− cells were then stained with peridinin-chlorophyll protein/Cy5.5-CD117 (BD), phycoerythrin (PE)/Cy7-Sca-1 (BioLegend), Alexa Fluor 700–CD48 (BioLegend) and PE-CD150 (BioLegend), allophycocyanin (APC)/Cy7-CD16/32 (FcR) (BioLegend), APC-CD105 (BioLegend). HSPC populations were identified as follows: long-term HSCs, Lin−Kit+Sca1+ (LKS+) CD48−CD150+; multipotent progenitors (MPPs), LKS+CD48+CD150−; granulocyte macrophage progenitors (GMPs), Lin−Kit+Sca1− (LKS−) CD150−FcR−; pre-GMP, LKS−CD150+CD105−; pre-Mk erythroid progenitors (MEPs), LKS−CD150+FcR−CD105−; pre-colony-forming unit (pre-CFU) erythroid, LKS−CD150+FcR−CD105+; erythroid progenitors, LKS−CD150−FcR−CD105+.18
To analyze cell cycle or pSmad2/3 staining in HSPCs, lineage-depleted cells were stained with streptavidin (BioLegend), APC-CD117 (BD), and PE/Cy7-Sca-1 (BioLegend), Alexa Fluor 700–CD48 (BioLegend), and PE or BV605-CD150 (BioLegend) and then fixed and permeabilized using CytoFix/Perm (BD). For cell cycle, surface stained, fixed, permeabilized cells were stained with peridinin-chlorophyll protein/Cy5.5-Ki67 (BD) and Hoechst 33342 (Invitrogen). To analyze the pSmad2/3 signal, fixed, permeabilized cells were stained first with pSmad2/3 primary antibody (Cell Signaling Technology) and then with secondary anti-rabbit, antibody Cy3 (The Jackson Laboratory).
Specificity of pSmad2/3 staining was tested by treating C57BL/6J mice with either the TGFβ-neutralizing antibody 1D11 (10 mg/kg) or a nontargeting control antibody 13C4 (10 mg/kg) on days 5, 10, and 15. Mice were euthanized on day 16 for analysis. Smad2/3 phosphorylation was assessed by intracellular flow cytometry as described previously.
Analysis gates were set based upon the fluorescence minus 1 (FMO) fluorophore. Multidimensional fluorescence-activated cell sorter analysis was performed using a BD LSRII equipped with 5 lasers. FlowJo (TreeStar) and FCS Express (De Novo) were used to analyze flow cytometry data.
Immunophenotypic analysis of EPs by flow cytometry
Mice were euthanized by CO2 asphyxiation and bones or spleen were processed as above except lineage depletion was not performed. Cell suspensions were stained with APC-conjugated Ter119, APC/Cy7-conjugated CD44, Hoechst and, in the indicated experiments, cells were stained with a modified biotinylated lineage cocktail (CD3, B220, CD11b and Gr1) and streptavidin. In the indicated experiments, cells were also stained with PE/Cy7-Kit (Abcam) and EPO receptor (Epor) primary antibody (R&D Systems) and then stained with secondary, anti-goat Cy3 antibody (The Jackson Laboratory).
Stem and progenitor cell assays
Primitive myeloid progenitors were enumerated using the spleen CFUs (CFU-S) after 12 days (CFU-S12) assay. Recipient mice were sublethally irradiated (7 Gy) using a 137Cs–γ-ray source and injected with 105 bone marrow (BM) cells. Spleens were isolated 12 days after transplantation and fixed in Bouin solution. The number of macroscopic spleen colonies was counted and expressed as the number of CFU-S colonies per 105 donor cells. Clonogenic myeloid progenitors were assessed by standard methylcellulose colony-forming cell (CFC) assays (MethoCult GF M3434; Stem Cell Technologies) using 1.5 × 104 BM mononuclear cells per well (6-well plate). Colonies were scored after 7 days of incubation and expressed as the number of macroscopic spleen colonies after 12 days (CFU-S12) per 1.5 × 104 BM mononuclear cells before normalizing to total leg (femur/tibia) cell counts.
HSC transplantation
Recipient mice were irradiated with 9 Gy using a 137Cs–γ-ray source. Three to 4 hours after irradiation, whole BM cells (marrow) from donor animals were infused via the tail vein of recipient animals. Competitive repopulation was used to assess the relative number of HSCs in marrow from TGFβ1ΔMk/ΔMk and TGFβ1FL/FL mice. TGFβ1ΔMk/ΔMk or TGFβ1FL/FL marrow cells were mixed with an equal number of marrow cells from congenic CD45.1+/+ control mice. The 1:1 (test/competitor) mixture (2 × 106 cells total) was injected into lethally irradiated mice via tail vein. Peripheral blood was analyzed for lymphoid (CD3/CD19) and myeloid (Gr1/CD11b) engraftment by flow cytometry at various times after transplantation. Animals were euthanized after 16 weeks and marrow harvested for immunophenotyping of HSPC populations and for serial transplantation. Serial transplantation of secondary recipients was performed as for primary recipients except for the marrow donor cells, which were not mixed with additional congenic competitor cells.
Cell count analysis
To assess blood count recovery, we collected peripheral blood (50 µL) into EDTA-coated capillary tubes (Thermo Fisher Scientific). Differential blood counts were measured using an automated ADVIA 120 Multispecies Hematology Analyzer (Bayer HealthCare) calibrated for murine blood.
Apoptosis analysis by flow cytometry
Apoptosis in mononuclear cell suspensions was assessed using the Annexin V-FITC Apoptosis Kit (BioLegend) according to manufacturer’s instructions and analyzed via flow cytometry. In some experiments, apoptosis was assessed using CellEvent Caspase-3/7 Green Detection Reagent by incubating marrow cells for 10 minutes prior to analysis by flow cytometry. Cleaved Caspase 3 was also assessed by intracellular staining of surface-stained mononuclear cell suspensions after permeabilization using CytoFix/Perm (BD) and subsequent staining with cleaved Caspase 3 primary antibody (Cell Signaling Technology) and anti-rabbit secondary antibody Cy3 (The Jackson Laboratory).
Immunofluorescence
Femurs were fixed in 4% paraformaldehyde overnight, and then decalcified using 10% EDTA before freezing at optimal cutting temperature (Sakura Finetek). Immunofluorescence staining was performed on frozen sections. After blocking with blocking buffer (3% bovine serum albumin, 3% serum, and 0.03% Tween-20), sections were incubated in primary antibodies overnight at 4°C using anti-cleaved Caspase 3 (Cell Signaling Technology) antibody. After washing, sections were stained with secondary antibody (anti-rabbit) CY3 (The Jackson Laboratory). The specificity of staining was confirmed in sequential sections using the secondary antibody alone. Images were acquired using a Zeiss spinning disk confocal microscope or a Zeiss 710 laser-scanning confocal microscope.
In vivo stimulation of erythropoiesis
Human recombinant EPO (300 U/kg body weight, ∼9 U per mouse) was injected subcutaneously for 5 consecutive days. On day 6, blood was sampled and mice were euthanized for analysis.
To induce hemolysis, mice were injected intraperitoneally with phenylhydrazine hydrochloride at 60 mg/kg (Sigma-Aldrich) on days 1 and 2. Blood was sampled on days 3, 5, 8, and 14 from staggered cohorts of mice.
To assess the ability of exogenous TGFβ1 to rescue the megakaryocytic TGFβ1 knockout, mice were treated with TGFβ1 (5 μg/kg per day) subcutaneously for 5 consecutive days as indicated. On day 6, blood was sampled and mice were euthanized for analysis.
Immunoblotting
Mononuclear cell suspensions were washed in PBS, pelleted, and lysed in lysis buffer (50 mM Tris-HCl [pH 7.5], 120 mM NaCl, and 0.4% NP-40 supplemented with proteinase inhibitors; samples were transferred to polyvinylidene difluoride [PVDF] membranes [EMD Millipore]) and blocked with 5% nonfat dried milk in PBS with 0.1% Tween 20. Primary and secondary antibodies were diluted in blocking solution. Primary antibodies against cleaved Caspase 3 (Cell Signaling Technology) were used. Secondary peroxidase-conjugated anti-rabbit antibody (EMD Millipore) was used before chemiluminescent visualization using the SuperSignal West Femto Substrate (Thermo Fisher Scientific).
Statistical analysis
All data are expressed as mean plus or minus standard deviation (SD). The Student t test (2-tailed) was used to analyze the statistical differences between groups, with the P values indicated in the plots (*P < .05, **P < .01, ***P < .001, or if not shown, the comparison was not significant).
Results
Conditional deletion of TGFβ1 in Mks increases BM apoptosis
Many HSCs reside in close proximity to Mks where they can be maintained in a quiescent state by niche signals such as TGFβ1 and CXCL4.19,20 Although genetic ablation of Mks using an inducible diphtheria toxin receptor under control of Mk-specific Cxcl4-cre driver releases HSCs from quiescence and licenses expansion of the HSC pool, it is not known how this affects blood cell production or other aspects of hematopoiesis. We selectively deleted Tgfb1 (TGFβ1) in Mks (TGFβ1ΔMk/ΔMk) and found that peripheral blood counts were entirely normal in TGFβ1ΔMk/ΔMk mice compared with TGFβ1FL/FL littermate controls despite the pool of primitive hematopoietic cells being expanded (Figure 1A). Sequestration of maturing cells within hematopoietic tissues could not explain this discrepancy because total BM and spleen cellularity were normal in TGFβ1ΔMk/ΔMk mice (Figure 1B). Excess HSCs in TGFβ1ΔMk/ΔMk mice appeared capable of robust differentiation because the number of immature Lin− hematopoietic progenitor cells (HPCs) was increased in the marrows of TGFβ1ΔMk/ΔMk mice (Figure 1C). Thus, it remained unexplained why the expanded number of HSCs and HPCs did not increase blood counts and marrow cellularity.
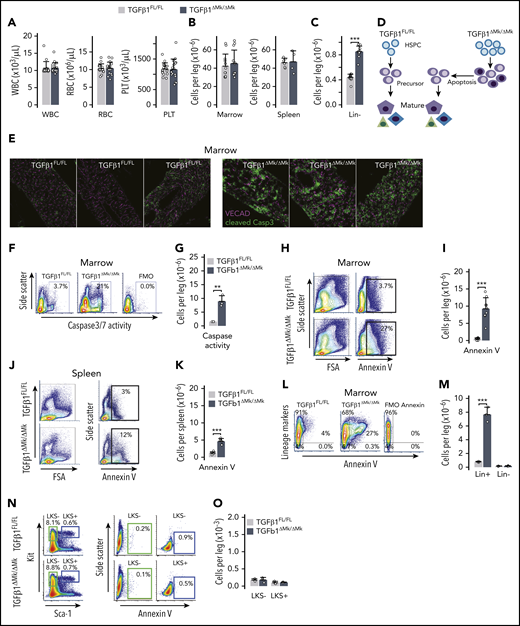
Conditional deletion of TGFβ1 in Mks increases apoptosis. (A) White blood cell (WBC), RBC, and platelet (PLT) counts are shown for TGFβ1FL/FL and TGFβ1ΔMk/ΔMk mice (n = 16 per group). (B) Marrow (n = 12 per group) and spleen (n = 5 per group) cellularity are shown for TGFβ1FL/FL (gray) and TGFβ1ΔMk/ΔMk mice (black). (C) The number of Lin− (those not expressing mature lineage markers CD3, B220, CD11b, Gr1, or Ter119) marrow cells per leg (femur and tibia) is shown (n = 13 per group). (D) Schema showing how excess immature HSPCs may yield normal marrow cellularity and blood cell counts due to apoptosis of surplus hematopoietic precursors. (E) Confocal immunofluorescence imaging of cleaved Caspase 3 (Casp3; green, Cy3) and Cdh5 (magenta, Alexa Fluor 647) in BM sections of TGFβ1FL/FL and TGFβ1ΔMk/ΔMk mice (n = 3 per group). Original magnification ×20. (F) Representative flow cytometry data of cleaved Caspase 3/7 activity is shown for TGFβ1FL/FL and TGFβ1ΔMk/ΔMk mice and for the FMO− control. (G) Quantification of apoptotic cells was assessed using the Caspase 3/7 reporter (n = 3 per group). (H) Representative flow cytometry data showing annexin V staining in marrow from TGFβ1FL/FL (top row) and TGFβ1ΔMk/ΔMk (bottom row) mice. (I) Quantification of the number of annexin V+ apoptotic cells in the marrow of TGFβ1FL/FL and TGFβ1ΔMk/ΔMk mice (n = 9 per group). (J) Representative data for spleen, as shown for marrow in panel H. (K) Quantification of the number of annexin V+ apoptotic cells in the spleen of TGFβ1FL/FL and TGFβ1ΔMk/ΔMk mice (n = 6 per group). (L) Representative flow cytometry data showing lineage markers (Lin = CD3, B220, CD11b, Gr1, or Ter119) plotted against annexin V in marrow from TGFβ1FL/FL and TGFβ1ΔMk/ΔMk mice. (M) Quantification of the number of annexin V+ apoptotic Lin+ and Lin− cells in the marrow of TGFβ1FL/FL and TGFβ1ΔMk/ΔMk mice (n = 8). (N) Gating and representative flow cytometry data showing annexin V staining in LKS− and LKS+ marrow populations for TGFβ1FL/FL (top row) and TGFβ1ΔMk/ΔMk (bottom row) mice. (O) Quantification of the number of annexin V+ apoptotic LKS− and LKS+ cells in the marrow of TGFβ1FL/FL and TGFβ1ΔMk/ΔMk mice (n = 3 per group). All the quantified data are shown as mean plus or minus standard deviation (SD) (*P < .05, **P < .01, ***P < .001, or if not shown, the comparison was not significant). FSA, forward scatter area; TNC, total nucleated cell; VECAD, vascular endothelial cadherin.
Conditional deletion of TGFβ1 in Mks increases apoptosis. (A) White blood cell (WBC), RBC, and platelet (PLT) counts are shown for TGFβ1FL/FL and TGFβ1ΔMk/ΔMk mice (n = 16 per group). (B) Marrow (n = 12 per group) and spleen (n = 5 per group) cellularity are shown for TGFβ1FL/FL (gray) and TGFβ1ΔMk/ΔMk mice (black). (C) The number of Lin− (those not expressing mature lineage markers CD3, B220, CD11b, Gr1, or Ter119) marrow cells per leg (femur and tibia) is shown (n = 13 per group). (D) Schema showing how excess immature HSPCs may yield normal marrow cellularity and blood cell counts due to apoptosis of surplus hematopoietic precursors. (E) Confocal immunofluorescence imaging of cleaved Caspase 3 (Casp3; green, Cy3) and Cdh5 (magenta, Alexa Fluor 647) in BM sections of TGFβ1FL/FL and TGFβ1ΔMk/ΔMk mice (n = 3 per group). Original magnification ×20. (F) Representative flow cytometry data of cleaved Caspase 3/7 activity is shown for TGFβ1FL/FL and TGFβ1ΔMk/ΔMk mice and for the FMO− control. (G) Quantification of apoptotic cells was assessed using the Caspase 3/7 reporter (n = 3 per group). (H) Representative flow cytometry data showing annexin V staining in marrow from TGFβ1FL/FL (top row) and TGFβ1ΔMk/ΔMk (bottom row) mice. (I) Quantification of the number of annexin V+ apoptotic cells in the marrow of TGFβ1FL/FL and TGFβ1ΔMk/ΔMk mice (n = 9 per group). (J) Representative data for spleen, as shown for marrow in panel H. (K) Quantification of the number of annexin V+ apoptotic cells in the spleen of TGFβ1FL/FL and TGFβ1ΔMk/ΔMk mice (n = 6 per group). (L) Representative flow cytometry data showing lineage markers (Lin = CD3, B220, CD11b, Gr1, or Ter119) plotted against annexin V in marrow from TGFβ1FL/FL and TGFβ1ΔMk/ΔMk mice. (M) Quantification of the number of annexin V+ apoptotic Lin+ and Lin− cells in the marrow of TGFβ1FL/FL and TGFβ1ΔMk/ΔMk mice (n = 8). (N) Gating and representative flow cytometry data showing annexin V staining in LKS− and LKS+ marrow populations for TGFβ1FL/FL (top row) and TGFβ1ΔMk/ΔMk (bottom row) mice. (O) Quantification of the number of annexin V+ apoptotic LKS− and LKS+ cells in the marrow of TGFβ1FL/FL and TGFβ1ΔMk/ΔMk mice (n = 3 per group). All the quantified data are shown as mean plus or minus standard deviation (SD) (*P < .05, **P < .01, ***P < .001, or if not shown, the comparison was not significant). FSA, forward scatter area; TNC, total nucleated cell; VECAD, vascular endothelial cadherin.
Hematopoietic cell population size is determined by the balance of cell gain (proliferation/self-renewal and differentiation) and cell loss (apoptosis and differentiation). Many late-acting hematopoietic cytokines are not required for lineage commitment yet provide essential proliferation, differentiation, and survival signals during maturation of hematopoietic precursors.6,21 Thus, we hypothesized that the excess progenitors observed in the TGFβ1ΔMk/ΔMk mice failed to increase blood counts because their progeny were unneeded, and inadequately supported by homeostatic levels of late-acting cytokines (Figure 1D). Indeed, BM apoptosis was markedly increased in the TGFβ1ΔMk/ΔMk mice compared with controls, as reported by cleaved Caspase 3 (Figure 1E-G; supplemental Figure 1a-b, available on the Blood Web site) or annexin V detection (Figure 1H-K). The excess apoptotic cells seemed largely restricted to hematopoietic cells expressing the lineage markers (Lin) Gr1, CD11b, CD3, B220, or Ter119 (Figure 1L-M). Annexin V staining of LKS− HPCs and LKS+ HSPCs was rare in both TGFβ1ΔMk/ΔMk mice and littermate controls (Figure 1N-O). These results support an interpretation that excess, unneeded hematopoietic precursors are pruned by apoptosis during hematopoietic differentiation.
Increased HSPCs in TGFβ1ΔMk/ΔMk mice have normal function
To rule out the possibility that progeny of HSPCs from TGFβ1ΔMk/ΔMk mice are intrinsically defective, we performed competitive repopulation assays by cotransplanting BM from TGFβ1ΔMk/ΔMk mice or TGFβ1FL/FL littermates competitively with CD45.1 congenic donor cells. As expected, TGFβ1FL/FLdonor cells engrafted and contributed to multilineage hematopoiesis indistinguishably from the congenic CD45.1 controls (Figure 2A; supplemental Figure 1d-f). In contrast, TGFβ1ΔMk/ΔMk donor cells outcompeted control cells, confirming that TGFβ1ΔMk/ΔMk donors are enriched for functional HSCs and indicating that these TGFβ1ΔMk/ΔMk HSCs yield progeny fully capable of reconstituting hematopoiesis after transplant. Serial transplantation of recipients demonstrated HSC self-renewal and contribution to hematopoiesis was intrinsically normal in the TGFβ1ΔMk/ΔMk donor cells (Figure 2B; supplemental Figure 1g-h).
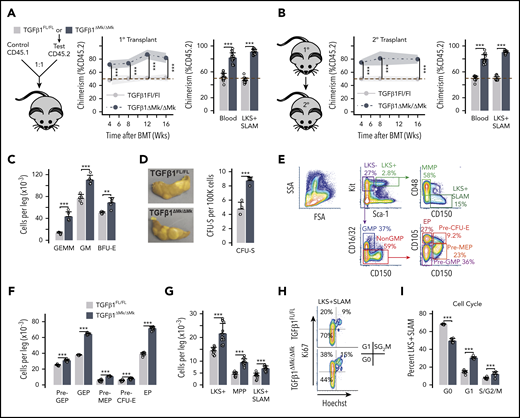
Increased HSPCs in TGFβ1ΔMk/ΔMk mice have normal function. (A) Schematic for the competitive repopulation assay is shown. Marrow CD45.2 donor cells from either TGFβ1FL/FL or TGFβ1ΔMk/ΔMk mice were mixed in a 1:1 ratio with marrow donor cells from congenic CD45.1 mice, then transplanted into lethally irradiated mice (n = 8 per group). CD45.2 chimerism in peripheral blood is shown at the indicated times after transplant (left). Chimerism of blood and BM LKS+ signaling lymphocyte activation molecule (SLAM; Lin−, Kit+, Sca1+, CD150+, CD48−) cells is shown 16 weeks after transplant for mice having received TGFβ1FL/FL (gray) or TGFβ1ΔMk/ΔMk (black) donor cell grafts, as assessed by flow cytometry. (B) Schematic for secondary transplantation of marrow harvested from primary recipients is shown. CD45.2 chimerism in peripheral blood is shown at the indicated times after secondary transplant (left). Chimerism in blood and in peripheral blood and marrow LKS+SLAM is shown 16 weeks after secondary transplant as for panel A (16 mice were transplanted in 2 independent experiments; 8 mice were transplanted per group in each experiment). (C) CFC enumeration of functional myeloid HPCs is shown for TGFβ1FL/FL and TGFβ1ΔMk/ΔMk marrow cells (n = 5 per group). The total number of CFU granulocyte, erythroid, monocyte, Mk progenitors (CFU-GEMM), CFU granulocyte macrophage (CFU-GM), and burst-forming unit erythroid progenitors (BFU-E) is shown per leg (femur and tibia). (D) The number of spleen CFUs (CFU-S12) 12 days after transplantation is shown per 100 000 donor cells transplanted (n = 4 per group). (E) Gating and representative flow cytometry data are shown for the indicated HSPC populations. (F) Quantification of the number of marrow cells with the immunophenotype of common myeloid progenitor (CMP), GMP, MEP, erythroid progenitors (EP), pre-GMP, pre-MEP is shown for TGFβ1FL/FL and TGFβ1ΔMk/ΔMk mice (n = 12 per group). (G) The number of LKS+ (Lin−Kit+Sca1+), MPP and LKS+SLAM cells per leg is shown for TGFβ1FL/FL and TGFβ1ΔMk/ΔMk mice (n = 12 per group). (H) Gating and representative flow cytometry data are shown for LKS+SLAM cell cycle as reported by Ki67 and Hoechst staining. (I) Cell cycle of BM LKS+SLAM HSCs is shown for TGFβ1FL/FL and TGFβ1ΔMk/ΔMk (mice n = 6 per group). All of the quantified data are shown as mean plus or minus SD (*P < .05, **P < .01, ***P < .001, or if not shown, the comparison was not significant). BMT, BM transplantation; SSA, side scatter area.
Increased HSPCs in TGFβ1ΔMk/ΔMk mice have normal function. (A) Schematic for the competitive repopulation assay is shown. Marrow CD45.2 donor cells from either TGFβ1FL/FL or TGFβ1ΔMk/ΔMk mice were mixed in a 1:1 ratio with marrow donor cells from congenic CD45.1 mice, then transplanted into lethally irradiated mice (n = 8 per group). CD45.2 chimerism in peripheral blood is shown at the indicated times after transplant (left). Chimerism of blood and BM LKS+ signaling lymphocyte activation molecule (SLAM; Lin−, Kit+, Sca1+, CD150+, CD48−) cells is shown 16 weeks after transplant for mice having received TGFβ1FL/FL (gray) or TGFβ1ΔMk/ΔMk (black) donor cell grafts, as assessed by flow cytometry. (B) Schematic for secondary transplantation of marrow harvested from primary recipients is shown. CD45.2 chimerism in peripheral blood is shown at the indicated times after secondary transplant (left). Chimerism in blood and in peripheral blood and marrow LKS+SLAM is shown 16 weeks after secondary transplant as for panel A (16 mice were transplanted in 2 independent experiments; 8 mice were transplanted per group in each experiment). (C) CFC enumeration of functional myeloid HPCs is shown for TGFβ1FL/FL and TGFβ1ΔMk/ΔMk marrow cells (n = 5 per group). The total number of CFU granulocyte, erythroid, monocyte, Mk progenitors (CFU-GEMM), CFU granulocyte macrophage (CFU-GM), and burst-forming unit erythroid progenitors (BFU-E) is shown per leg (femur and tibia). (D) The number of spleen CFUs (CFU-S12) 12 days after transplantation is shown per 100 000 donor cells transplanted (n = 4 per group). (E) Gating and representative flow cytometry data are shown for the indicated HSPC populations. (F) Quantification of the number of marrow cells with the immunophenotype of common myeloid progenitor (CMP), GMP, MEP, erythroid progenitors (EP), pre-GMP, pre-MEP is shown for TGFβ1FL/FL and TGFβ1ΔMk/ΔMk mice (n = 12 per group). (G) The number of LKS+ (Lin−Kit+Sca1+), MPP and LKS+SLAM cells per leg is shown for TGFβ1FL/FL and TGFβ1ΔMk/ΔMk mice (n = 12 per group). (H) Gating and representative flow cytometry data are shown for LKS+SLAM cell cycle as reported by Ki67 and Hoechst staining. (I) Cell cycle of BM LKS+SLAM HSCs is shown for TGFβ1FL/FL and TGFβ1ΔMk/ΔMk (mice n = 6 per group). All of the quantified data are shown as mean plus or minus SD (*P < .05, **P < .01, ***P < .001, or if not shown, the comparison was not significant). BMT, BM transplantation; SSA, side scatter area.
Ex vivo HPC function appeared normal in TGFβ1ΔMk/ΔMk mice, suggesting that these cells were not intrinsically defective in TGFβ1ΔMk/ΔMk mice (Figure 2C). The number of GMPs, burst-forming unit erythroid progenitors (BFU-E) and CFU granulocyte, erythroid, monocyte, Mk progenitors (CFU-GEMM) were all increased in the marrow of TGFβ1ΔMk/ΔMk mice as reported by CFC assays. Similarly, the CFU-S12 assay showed a greater number of functionally normal MPPs in TGFβ1ΔMk/ΔMk donor marrow compared with TGFβ1FL/FL littermate controls (Figure 2D). Quantified HSPC immunophenotypes correlated well with functional measures, again demonstrating that the number of HSPCs is increased in TGFβ1ΔMk/ΔMk mice (Figure 2E-G). The larger number of HSPCs and their more proliferative character in TGFβ1ΔMk/ΔMk mice (Figure 2H-I) indicate that megakaryocytic TGFβ1 normally curbs the HSPC pool size. Yet, progeny of these excess HSPCs appear to undergo apoptosis rather than contribute to BM cellularity and mature blood cell counts.
Excess erythroid precursors undergo apoptosis in vivo
The islands of apoptotic cells observed in BM of TGFβ1ΔMk/ΔMk mice (Figure 1E) are reminiscent of erythroid islands, suggesting that excess EPs may be contributing to the formation of these clusters. Indeed, the proportion of annexin V–staining apoptotic Ter119+ EP cells (EPs) was approximately sixfold higher in the marrow and spleen of TGFβ1ΔMk/ΔMk mice compared with TGFβ1FL/FL littermate controls (Figure 3A-B). We quantified apoptosis within well-defined populations of maturing erythroid precursors22-24 (Figure 3C) using annexin V. Although the number of immature EPs, such as pro/basophilic (EI), polychromatophilic (EII), and orthochromatophilic erythroblasts (EIII), was increased in TGFβ1ΔMk/ΔMk mice, the excess seemed largely composed of cells staining for the apoptotic marker annexin V (Figure 3D). In contrast, the number of mature reticulocytes and erythrocytes (EIV/EV) in TGFβ1ΔMk/ΔMk mice was indistinguishable from TGFβ1FL/FL. Thus, the excess erythroid committed precursors are culled during early maturation, thereby producing normal numbers of erythrocytes and normal RBC counts.
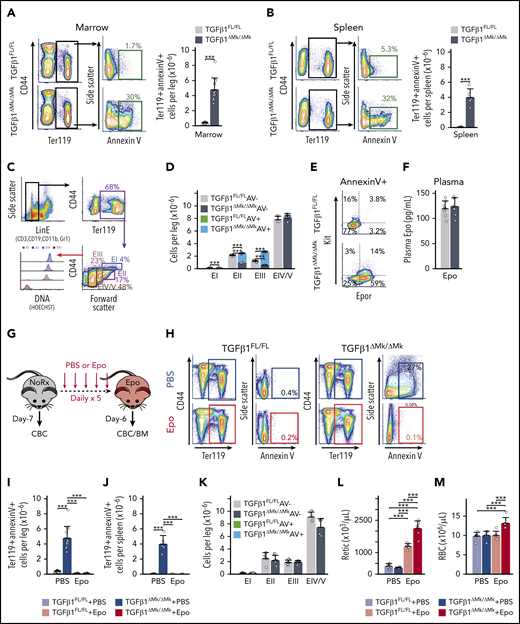
Surplus EPO-dependent EPs undergo apoptosis in vivo. (A) Gating and representative flow cytometry data are shown for annexin V staining of Ter119+ EPs in marrow from TGFβ1FL/FL (top row) and TGFβ1ΔMk/ΔMk (bottom row) mice. Quantification of the number of marrow, annexin V staining, apoptotic Ter119+ erythroid cells is shown for TGFβ1FL/FL and TGFβ1ΔMk/ΔMk mice (n = 12 per group). (B) Gating and representative flow cytometry data are shown spleen cells as described for panel A. Quantification of splenic apoptotic Ter119+ erythroid cells is shown for TGFβ1FL/FL and TGFβ1ΔMk/ΔMk mice (n = 4-6 per group). (C) Flow cytometry gating strategy using Lin− erythroid LinE− (cells not expressing CD3, B220, Gr1, or CD11b), CD44, Ter119, Hoechst (DNA), and annexin V to characterize erythroid maturation. (D) Quantification of EPs is shown (n = 3 per group). Viable cells and annexin V+ apoptotic cells for TGFβ1FL/FL (gray/green) and TGFβ1ΔMk/ΔMk (black/blue) mice, respectively. (E) Representative flow cytometry showing staining of Kit and Epor within the annexin V+/Ter119+ population identifies excess Epor+/Kit− erythroblasts within the TGFβ1ΔMk/ΔMk marrow. (F) Plasma EPO levels are shown as assessed by enzyme-linked immunosorbent assay (ELISA; n = 8 per group). (G) TGFβ1FL/FL and TGFβ1ΔMk/ΔMk mice were treated with 300 U/kg Epo daily for 5 days and euthanized on day 6 for analysis. (H) Representative annexin V staining of marrow EPCs is shown after PBS (top) or Epo (bottom) treatment of TGFβ1FL/FL (left) and TGFβ1ΔMk/ΔMk (right) mice. The number of annexin V+ apoptotic Ter119+ EPCs is shown in marrow (I) and spleen (J) before and after EPO treatment (n = 4-12 per group). (K) Apoptosis within maturing EPs is shown for marrow cells after EPO treatment, as shown in panel D. Viable cells and annexin V+ apoptotic cells for Epo-treated TGFβ1FL/FL (pink/green) and TGFβ1ΔMk/ΔMk (red/blue) mice, respectively (n = 4 per group). Reticulocyte (Retic) counts (L) and RBCs (M) are shown for TGFβ1FL/FL (gray/pink) and TGFβ1ΔMk/ΔMk (black/red) mice before and after Epo (n = 6 per group). All of the quantified data are shown as mean plus or minus SD (*P < .05, **P < .01, ***P < .001, or if not shown, the comparison was not significant). AV, annexin V; CBC, complete blood count; Rx, prescription.
Surplus EPO-dependent EPs undergo apoptosis in vivo. (A) Gating and representative flow cytometry data are shown for annexin V staining of Ter119+ EPs in marrow from TGFβ1FL/FL (top row) and TGFβ1ΔMk/ΔMk (bottom row) mice. Quantification of the number of marrow, annexin V staining, apoptotic Ter119+ erythroid cells is shown for TGFβ1FL/FL and TGFβ1ΔMk/ΔMk mice (n = 12 per group). (B) Gating and representative flow cytometry data are shown spleen cells as described for panel A. Quantification of splenic apoptotic Ter119+ erythroid cells is shown for TGFβ1FL/FL and TGFβ1ΔMk/ΔMk mice (n = 4-6 per group). (C) Flow cytometry gating strategy using Lin− erythroid LinE− (cells not expressing CD3, B220, Gr1, or CD11b), CD44, Ter119, Hoechst (DNA), and annexin V to characterize erythroid maturation. (D) Quantification of EPs is shown (n = 3 per group). Viable cells and annexin V+ apoptotic cells for TGFβ1FL/FL (gray/green) and TGFβ1ΔMk/ΔMk (black/blue) mice, respectively. (E) Representative flow cytometry showing staining of Kit and Epor within the annexin V+/Ter119+ population identifies excess Epor+/Kit− erythroblasts within the TGFβ1ΔMk/ΔMk marrow. (F) Plasma EPO levels are shown as assessed by enzyme-linked immunosorbent assay (ELISA; n = 8 per group). (G) TGFβ1FL/FL and TGFβ1ΔMk/ΔMk mice were treated with 300 U/kg Epo daily for 5 days and euthanized on day 6 for analysis. (H) Representative annexin V staining of marrow EPCs is shown after PBS (top) or Epo (bottom) treatment of TGFβ1FL/FL (left) and TGFβ1ΔMk/ΔMk (right) mice. The number of annexin V+ apoptotic Ter119+ EPCs is shown in marrow (I) and spleen (J) before and after EPO treatment (n = 4-12 per group). (K) Apoptosis within maturing EPs is shown for marrow cells after EPO treatment, as shown in panel D. Viable cells and annexin V+ apoptotic cells for Epo-treated TGFβ1FL/FL (pink/green) and TGFβ1ΔMk/ΔMk (red/blue) mice, respectively (n = 4 per group). Reticulocyte (Retic) counts (L) and RBCs (M) are shown for TGFβ1FL/FL (gray/pink) and TGFβ1ΔMk/ΔMk (black/red) mice before and after Epo (n = 6 per group). All of the quantified data are shown as mean plus or minus SD (*P < .05, **P < .01, ***P < .001, or if not shown, the comparison was not significant). AV, annexin V; CBC, complete blood count; Rx, prescription.
Excess EPs are rescued by exogenous EPO
Apoptotic Ter119+ EPs within TGFβ1ΔMk/ΔMk marrow predominantly expressed the Epor but not Kit (Figure 3E). EPO provides a survival signal to Epor+ EPs, allowing them to escape apoptosis and continue differentiation.8-10 Excess apoptosis of EPs in TGFβ1ΔMk/ΔMk mice was not due to subnormal plasma EPO levels (Figure 3F). We reasoned that the surplus, unneeded Epor+ cells may not be supported by physiologic EPO levels. To test this, we treated mice with exogenous EPO (300 U/kg) (Figure 3G). Strikingly, exogenous Epo rescued the excess apoptosis of EPs in TGFβ1ΔMk/ΔMk marrow and spleen (Figure 3H-K). The erythropoietic response of TGFβ1ΔMk/ΔMk mice to Epo was much more robust compared with TGFβ1FL/FL littermate controls (Figure 3L-M), suggesting that the rescued apoptosis resulted in increased erythropoietic output. Using phenylhydrazine hydrochloride–induced hemolysis as an anemia model, we found that the erythropoietic response of TGFβ1ΔMk/ΔMk mice was much brisker and more robust than that of TGFβ1FL/FL littermate controls (supplemental Figure 2). These results demonstrate that, although genetic deletion of TGFβ1 in MKs expands the number of committed erythroid progenitors, the resultant glut of their EP progeny requires nonhomeostatic Epo levels to promote survival, expansion, and maturation to erythrocytes.
Mks are the dominant source of TGFβ1 signaling in HSPCs
Megakaryocytic TGFβ1 is necessary for robust phosphorylation of receptor-activated Smad2 and Smad3 (Smad2/3) in HSPCs, as assessed by flow cytometry with intracellular staining (Figure 4A). In contrast, we detected little phospho-Smad2/3 (pSmad2/3) in EPs in either the TGFβ1ΔMk/ΔMk mice or TGFβ1FL/FL littermate controls (Figure 4A). HSPCs in TGFβ1ΔMk/ΔMk mice were capable of normal TGFβ1 signaling because exogenous TGFβ1 induced strong phosphorylation of Smad2/3, restoring the mean fluorescence intensity (MFI) to the level found in TGFβ1FL/FL controls (Figure 4B-C). Genetic deletion of Tgfb1 in Schwann cells had no identifiable effect on BM cellularity, HSPC numbers, or HSC cell cycle (supplemental Figure 3). Thus, Mks serve as the major source of TGFβ triggering Smad2/3 phosphorylation in HSPCs.
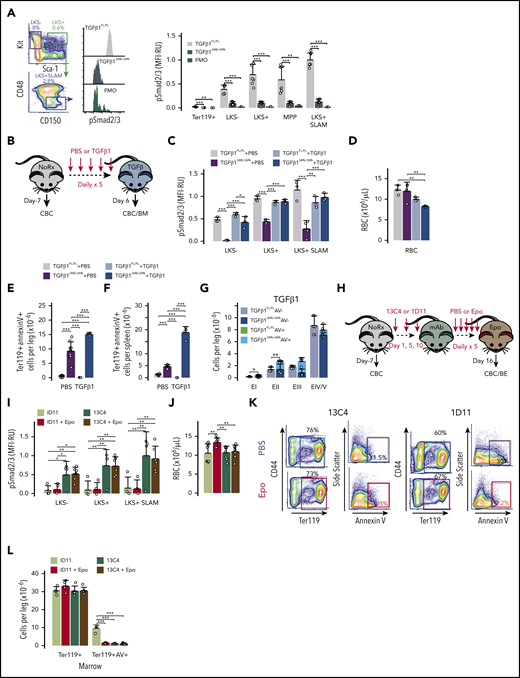
Mks direct TGFβ1 signaling in HSPCs but EP maturation is independently regulated. (A) Gating and representative flow cytometry data assessing Smad2/3 phosphorylation (pSmad2/3) within the LKS+SLAM population is shown for TGFβ1FL/FL and TGFβ1ΔMk/ΔMk mice and for the FMO− control. Mean fluorescence intensity (MFI) of pSmad2/3 within the indicated hematopoietic populations is shown for TGFβ1FL/FL and TGFβ1ΔMk/ΔMk mice (n = 7 per group). (B) Schematic showing TGFβ1FL/FL and TGFβ1ΔMk/ΔMk mice treated with 5 μg/kg TGFβ1 daily for 5 days and euthanized on day 6 for analysis. (C) MFI of pSmad2/3 staining is shown for Lin− HSPC subpopulations as quantified by flow cytometry of TGFβ1FL/FL (gray/light blue) and TGFβ1ΔMk/ΔMk (black/dark blue) mice treated with PBS or TGFβ1 (n = 3-4 mice per group). (D) RBC counts are shown for TGFβ1FL/FL and TGFβ1ΔMk/ΔMk mice treated with PBS or TGFβ1 (n = 6-7 per group). The number of Ter119+annexin V+ apoptotic cells in marrow (E) and spleen (F) is shown for TGFβ1FL/FL or TGFβ1ΔMk/ΔMk mice (n = 4-9 per group). (G) Apoptosis within maturing EPs is shown for marrow cells after TGFβ1 treatment (n = 3-4 per group), as shown in Figure 3D. (H) Schematic showing C57BL/6J mice treated with either a TGFβ-neutralizing antibody (1D11) or isotype control antibody (13C4) at a dose of 10 mg/kg on days 1, 5, and 10 and then treated daily for 5 days with either PBS or EPO (300 U/kg). All mice were euthanized for analysis on day 16. (I) MFI of pSmad2/3 staining is shown for Lin− HSPC subpopulations as quantified by flow cytometry of mice treated with 13C4 control antibody (dark green/tan) or 1D11 (light green/red) and then PBS or TGFβ1 (n = 4 to 6 mice per group). (J) RBC counts are shown for mice treated with 13C4 or 1D11 followed by either PBS or EPO (n = 8 per group). (K) Representative data for apoptosis within maturing EPs is shown for marrow cells for B6 mice treated with 13C4 or 1D11 and then either PBS or EPO, as shown in Figure 3D. (L) The total number Ter119+ and the number of annexin V+ apoptotic Ter119+ EPs is shown for mice treated with 13C4 or 1D11 and then either PBS or Epo (n = 4-6 per group). All of the quantified data are shown as mean plus or minus SD (*P < .05, **P < .01, ***P < .001, or if not shown, the comparison was not significant). mAb, monoclonal antibody; MFI-RU, MFI relative fluorescence units.
Mks direct TGFβ1 signaling in HSPCs but EP maturation is independently regulated. (A) Gating and representative flow cytometry data assessing Smad2/3 phosphorylation (pSmad2/3) within the LKS+SLAM population is shown for TGFβ1FL/FL and TGFβ1ΔMk/ΔMk mice and for the FMO− control. Mean fluorescence intensity (MFI) of pSmad2/3 within the indicated hematopoietic populations is shown for TGFβ1FL/FL and TGFβ1ΔMk/ΔMk mice (n = 7 per group). (B) Schematic showing TGFβ1FL/FL and TGFβ1ΔMk/ΔMk mice treated with 5 μg/kg TGFβ1 daily for 5 days and euthanized on day 6 for analysis. (C) MFI of pSmad2/3 staining is shown for Lin− HSPC subpopulations as quantified by flow cytometry of TGFβ1FL/FL (gray/light blue) and TGFβ1ΔMk/ΔMk (black/dark blue) mice treated with PBS or TGFβ1 (n = 3-4 mice per group). (D) RBC counts are shown for TGFβ1FL/FL and TGFβ1ΔMk/ΔMk mice treated with PBS or TGFβ1 (n = 6-7 per group). The number of Ter119+annexin V+ apoptotic cells in marrow (E) and spleen (F) is shown for TGFβ1FL/FL or TGFβ1ΔMk/ΔMk mice (n = 4-9 per group). (G) Apoptosis within maturing EPs is shown for marrow cells after TGFβ1 treatment (n = 3-4 per group), as shown in Figure 3D. (H) Schematic showing C57BL/6J mice treated with either a TGFβ-neutralizing antibody (1D11) or isotype control antibody (13C4) at a dose of 10 mg/kg on days 1, 5, and 10 and then treated daily for 5 days with either PBS or EPO (300 U/kg). All mice were euthanized for analysis on day 16. (I) MFI of pSmad2/3 staining is shown for Lin− HSPC subpopulations as quantified by flow cytometry of mice treated with 13C4 control antibody (dark green/tan) or 1D11 (light green/red) and then PBS or TGFβ1 (n = 4 to 6 mice per group). (J) RBC counts are shown for mice treated with 13C4 or 1D11 followed by either PBS or EPO (n = 8 per group). (K) Representative data for apoptosis within maturing EPs is shown for marrow cells for B6 mice treated with 13C4 or 1D11 and then either PBS or EPO, as shown in Figure 3D. (L) The total number Ter119+ and the number of annexin V+ apoptotic Ter119+ EPs is shown for mice treated with 13C4 or 1D11 and then either PBS or Epo (n = 4-6 per group). All of the quantified data are shown as mean plus or minus SD (*P < .05, **P < .01, ***P < .001, or if not shown, the comparison was not significant). mAb, monoclonal antibody; MFI-RU, MFI relative fluorescence units.
Exogenous TGFβ1 cannot rescue the erythroid phenotype of TGFβ1ΔMk/ΔMk mice
It is possible that TGFβ1 sensitizes EPs to EPO, and, if true, exogenous TGFβ1 should rescue EP dropout in TGFβ1ΔMk/ΔMk mice. To test this, we treated the mice with TGFβ1 for 5 days and assessed erythroid response (Figure 4B). Although exogenous TGFβ1 reestablished pSmad2/3 in TGFβ1ΔMk/ΔMk HSPCs (Figure 4C), it did not trigger an erythropoietic response. Rather, exogenous TGFβ1 induced mild anemia in the TGFβ1ΔMk/ΔMk mice (Figure 4D) coupled with worsened apoptosis in the marrow and spleen (Figure 4E-G). In contrast, exogenous TGFβ1 did not induce detectable changes in RBCs or apoptosis in TGFβ1FL/FL controls (Figure 4E-G). Although these studies cannot rule out functional interactions between TGFβ1 and EPO signaling during terminal erythropoiesis, they indicate that the excess erythroid apoptosis found in TGFβ1ΔMk/ΔMk mice does not directly result from insufficient Mk-derived TGFβ signaling in EPs. Thus, the effect of Mk TGFβ1 is restricted and predominantly acts on immature HSPCs.
TGFβ blockade stimulates overproduction of apoptotic erythroid-committed precursors that can be coupled to RBC production using low-dose EPO
Because we found that the Mks are responsible for the majority of TGFβ signaling in HSPCs, it is possible that blockade of TGFβ signaling could phenocopy these effects by inducing overproduction of erythroid-committed precursors. To test this, we pretreated C57BL6/J (B6) mice with a TGFβ1-neutralizing antibody (1D11) or nontargeting, isotype control antibody (13C4) and then either PBS or low-dose EPO (Figure 4H). TGFβ neutralization by 1D11 reduced pSmad2/3 MFI in HSPCs in wild-type mice whereas the 13C4 control had no effect, demonstrating on-target activity (Figure 4I). As we found in TGFβ1ΔMk/ΔMk mice, B6 mice treated with endogenous TGFβ neutralized by 1D11 had an expanded pool of Lin− and HSPCs in marrow compared with those receiving the control 13C4 antibody (supplemental Figure 4). Low-dose EPO triggered a brisk erythropoietic response in mice treated with 1D11 but not those treated with the 13C4 control (Figure 4J). Consistent with this finding, exogenous EPO rescued the EP dropout observed in B6 mice treated with 1D11 but did not affect the low apoptosis observed in mice treated with the 13C4 control (Figure 4K-L). Therefore, the boundary of megakaryocytic TGFβ1 activity is compartmentalized within the marrow with predominant effects on immature HSPCs while excluding their progeny (Figure 5).

Modular model of erythropoiesis compartmentalized by Mk TGFβ1. (A) Schematic of proposed compartmentalized model of erythropoiesis. Megakaryocytic TGFβ1 serves as a gatekeeper regulating the feed of committed erythroid progenitors to a maturation module regulated by EPO. EPO-dependent erythroblast survival is controlled by the need for RBC production sensed by oxygen delivery to renal EPO-producing cells. (B) Genetic deletion of TGFβ in Mks, or use of TGFβ ligand trap (1D11), licenses production of unneeded erythroid-committed progenitors. The excess EPs are not supported by homeostatic EPO, undergo apoptosis, and fail to contribute to RBC production. (C) Excess EPs can be rescued to exogenous EPO or increased physiologic demand (eg, supplemental Figure 2).
Modular model of erythropoiesis compartmentalized by Mk TGFβ1. (A) Schematic of proposed compartmentalized model of erythropoiesis. Megakaryocytic TGFβ1 serves as a gatekeeper regulating the feed of committed erythroid progenitors to a maturation module regulated by EPO. EPO-dependent erythroblast survival is controlled by the need for RBC production sensed by oxygen delivery to renal EPO-producing cells. (B) Genetic deletion of TGFβ in Mks, or use of TGFβ ligand trap (1D11), licenses production of unneeded erythroid-committed progenitors. The excess EPs are not supported by homeostatic EPO, undergo apoptosis, and fail to contribute to RBC production. (C) Excess EPs can be rescued to exogenous EPO or increased physiologic demand (eg, supplemental Figure 2).
Discussion
Erythropoiesis is subject to modular regulation. EPO acts during a very limited stage of differentiation, supporting early EPs as they gear up for iron accumulation, heme synthesis, and globin gene transcription.7,14,15 Accordingly, many causes of anemia are unresponsive to exogenous EPO. Recently, TGFβ superfamily activin receptor (Acvr2a/b) ligand traps have shown activity treating chronic EPO-unresponsive anemia in myelodysplastic syndrome and β-thalassemia, and are thought to promote erythroid maturation after EPO signaling.25-29 However, although activins can support erythropoiesis in vitro and in vivo,30,31 the molecular regulators of the EPO-unresponsive erythroid compartment are not well defined. Here, we show that the number of committed erythroid progenitors is indirectly controlled by the availability of megakaryocytic TGFβ1, adding a new level of erythroid regulation prior to the EPO-restriction point.
Megakaryocytic TGFβ1 helps to match the number of immature hematopoietic progenitor and stem cells to the production of mature effector cells. Deletion of Mk TGFβ leads to inefficient erythropoiesis because unneeded, excess EPs undergo apoptosis rather than contribute to blood cell output. Because Mk TGFβ1 ablation, or TGFβ blockade, broadly expands the pool of HSPCs, disruption of this pathway may disorder production of nonerythroid blood cell lines too. Direct testing will be required because the cytokines and maturation checkpoints controlling terminal granulopoiesis, thrombopoiesis, and lymphopoiesis differ substantially from erythropoiesis. Exogenous EPO or increased physiologic demand (supplemental Figure 2) rescued the excess EPs, leading to their brisk contribution to RBC counts. It is possible that other lineages could be similarly recruited from the enlarged pool of HSPCs if supported by appropriate cues. We previously found that TGFβ blockade during hematopoietic regeneration hastened multilineage reconstitution after chemotherapy, transplantation, or hemolysis.32 These results suggest that in the appropriate context, when cytokine levels are enhanced and the microenvironment is primed, the expanded pool of HSPCs can effectively contribute to blood cell production.
It is likely that normally the release and/or activation of latent Mk TGFβ is tightly controlled. Our data suggest that disrupting homeostatic levels of Mk TGFβ could be used to augment blood cell production, but HSC exhaustion is possible if this approach were used continuously, for long periods of time. Conversely, abnormally high Mk TGFβ signaling may also have potentially deleterious effects on hematopoiesis. Indeed, excessive, dysregulated TGFβ signaling is commonly implicated in the pathology of fibrogenic diseases such as pulmonary fibrosis and myelofibrosis,33-35 and excessive SMAD2/3 phosphorylation is found in the marrow of patients with myeloid malignancies36,37 (P.K. and J.M.S., unpublished observations in human myeloproliferative neoplasms). Intriguingly, genetic deletion of phosphatidylinositol transfer proteins in Mks leads to excess TGFβ secretion and myelosuppression.38 TGFβ is known to suppress erythropoiesis and megakaryopoiesis in vitro and in vivo,39,40 suggesting that aberrant Mk TGFβ could contribute to the anemia and thrombocytopenia that characterizes many myeloid malignancies. In this study, we found that exogenous TGFβ1 worsened the erythroid apoptosis and caused anemia in TGFβ1ΔMk/ΔMk mice. The biological effects of TGFβ1 differ based on ligand concentration, crosstalk with other signaling systems, and the type of downstream effector proteins used. For this reason, it can be reasonably expected that hematopoietic cells harboring pathogenic mutations could interpret TGFβ signals differently than normal HSPCs perhaps escaping some of the suppressive activities of this mediator. Elucidating the contributions of aberrant TGFβ signaling to abnormal hematopoiesis is a rich area for future research.
Although Mks and erythroids share a common progenitor, Mks also have a direct ontological link to a subset of Mk-biased HSCs.41 Understanding the evolutionary reasoning for Mks being placed at the helm of hematopoiesis and exploring the potential of manipulating this pathway clinically will be important.
For original data, please e-mail the corresponding author.
The online version of this article contains a data supplement.
The publication costs of this article were defrayed in part by page charge payment. Therefore, and solely to indicate this fact, this article is hereby marked “advertisement” in accordance with 18 USC section 1734.
Acknowledgments
This work was supported by grants from the National Institutes of Health, National Heart, Lung, and Blood Institute (HL1166436-01A1); TriSci (Tri-Sci 2013-030); and the Cancer Research and Treatment Fund (CR&T).
Authorship
Contribution: S.D.G. designed and performed experiments, and analyzed data, wrote the manuscript; P.K. performed experiments, analyzed data, and edited the manuscript; N. Mollé, M.M.Y., G.A.-Z., T.S., F.B., N. Messali, and R.S. performed experiments; S.R. reviewed data and edited the manuscript; and J.M.S. designed the research, analyzed the data, and wrote the manuscript.
Conflict-of-interest disclosure: The authors declare no competing financial interests.
Correspondence: Joseph M. Scandura, Weill Cornell Medicine, 1300 York Ave, Box 113, New York, NY 10065; e-mail: jms2003@med.cornell.edu.

This feature is available to Subscribers Only
Sign In or Create an Account Close Modal