

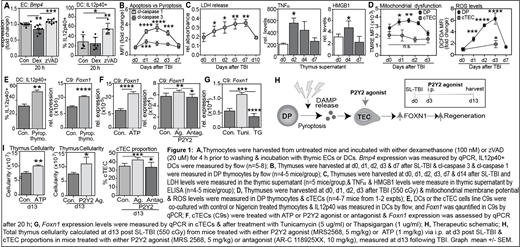
T cell reconstitution after transplant is critically dependent on the thymus; an inverse relationship between a transplant recipient's age and their capacity to generate T lymphocytes (in particular CD4+T cells) has been found in several studies, and thymic function pre-transplant can have a significant impact on clinical outcomes. Although the thymus has a remarkable ability to repair following damage, the mechanisms underlying this endogenous regeneration remain poorly understood. Despite this regenerative capacity, delayed T cell reconstitution is associated with an increased risk of infections, relapse of malignancy and the development of secondary malignancies. Therefore, there is a clinical demand for therapeutics that restore immune function after damage. Our recent studies have identified two key pathways driving thymic regeneration; centered on the secretion of BMP4 by endothelial cells (ECs) and IL-22 by innate lymphoid cells (Dudakov 2012 Science 336:91; Dudakov 2017 Blood130:933; Wertheimer 2018 Sci Immunol3:19). However, the specific regulatory mechanisms that trigger these regeneration-associated factors after damage remain unclear. Our previous work identified that the presence of homeostatic apoptotic CD4+CD8+ (DP) thymocytes, as apoptotic thymocytes form the bulk of developing T cells, suppress the production of IL-23 in dendritic cells (DCs), a key downstream mediator for IL-22, and BMP4 in ECs (Fig. 1A), and that the depletion of apoptotic thymocytes after damage precedes the production of these regenerative factors. Therefore, together with our findings that the metabolic needs of key thymus populations alter drastically following injury due to damage-induced metabolic remodeling, we hypothesized that further to the loss of DP-specific suppression, metabolic dysfunction in DPs after damage triggers mitochondrial-induced pyroptotic cell death, which can directly promote regeneration of the thymus.
Consistent with this hypothesis, our preliminary data shows increased levels of cl-caspase 1 (pyroptotic caspase) and a decrease in cl-caspase 3 (apoptotic caspase) in DPs after SL-TBI (550 cGy), demonstrating a preferential induction of pyroptotic cell death in DPs after damage (Fig. 1B). Furthermore, we demonstrated an increase in extracellular lactate dehydrogenase (LDH) levels, HMGB-1 and TNF⍺[canonical damage-associated molecular patterns (DAMPs) released during ICD] acutely after damage caused by SL-TBI (Fig. 1C).Given our previous findings that stromal cells are more radio-resistant than DP thymocytes (Wertheimer 2018 Sci Immunol3:19), and evidence for mitochondrial-induced pyroptosis, we identified hyperpolarization of the mitochondrial membrane potential accompanied by increased levels of ROS in DPs, an effect not observed in TECs, suggesting metabolic stability confers protection against acute damage (Fig. 1D). Furthermore, co-culture of pyroptotic thymocytes results in increased IL12p40+ DCs and increased Foxn1 expression in TECs (Fig. 1E), strengthening our hypothesis that cell-cell communication drives thymic regeneration after damage by inducing regenerative factors as well as directly promoting TEC function via secreted factors from pyroptotic DPs. One way in which DAMPs, such as ATP, can initiate cell signaling is by the activation of cell surface purinergic receptors, including P2Y2 which is widely expressed on TECs, and here we demonstrate that in vitro treatment with ATP or P2Y2 agonist increases Foxn1 in cTECs, and P2Y2 antagonism reverses this effect (Fig 1F). As P2Y2 activation promotes Ca2+efflux from the ER, we have further demonstrated that stimulating the intracellular release of Ca2+, using tunicamycin, induced Foxn1 expression in cTECs, which was reversed upon inhibition of Ca2+release (Fig. 1G). Importantly, we demonstrate here that this pathway can be therapeutically targeted by activating P2Y2 signaling in vivo with MRS2568 or ATP enhances thymus cellularity and expands cTECs in models of acute injury (Fig. 1H&I).
These findings not only reveal a novel metabolic-mediated molecular mechanism governing tissue regeneration; but also by targeting FOXN1 directly offers a potentially superior therapeutic strategy for boosting thymic regeneration and T cell reconstitution after damage such as that caused by HCT, infection or cytoreductive therapy.
No relevant conflicts of interest to declare.

This feature is available to Subscribers Only
Sign In or Create an Account Close Modal