

Introduction: Sickle cell anemia (SCA) is a severe and life-threatening disorder that requires treatment to prevent short- and long-term complications and prolong life. The primary disease-modifying therapy for SCA remains hydroxyurea (HU). Due to 30 years of evidence demonstrating safety and efficacy culminating with BABY HUG, the 2014 National Heart, Lung, and Blood Institute (NHLBI) guidelines recommended offering HU to all children with the severe sickle cell genotypes (HbSS, HbS-0thalassemia) beginning at 9 months of age. Despite these recommendations, HU utilization in pediatric patients in the US remains with rates reported as low as 38-47% among the most severe genotypes as recently as 2017. Providers have identified a number of barriers to more widespread use, including the inability to identify which patients may benefit, concern for possible side effects, uncertainties regarding dose, and concerns regarding possible nonadherence. As complications begin as early as the first year of life, it is a disservice to withhold a proven therapy. Here, we describe the effective and nearly universal uptake of HU in our pediatric SCA population at Cincinnati Children's Hospital Medical Center (CCHMC).
Methods: We performed an IRB-approved retrospective review to assess the hydroxyurea prescribing practices and clinical complications of patients with SCA treated at CCHMC from 2010-2019. Following the NHLBI guidelines' release in 2014, we changed the recommended age of HU initiation to be within the first year of life. Corresponding with this change, we have initiated HU for most young children with an individualized, pharmacokinetics (PK)-guided dosing strategy through both the Therapeutic Response Evaluation and Adherence Trial (TREAT, NCT03789591) and the Hydroxyurea Optimization through Precision Study (HOPS, NCT03789591). Due to the onset of symptoms for some patients before 9 months, we have offered HU initiation as early as 6 months of age since 2015.
Our objective was to compare the rates of HU utilization, age of initiation, and hospitalization rate before (2010-13) and after (2014-19) the release of the NHLBI guidelines and the start of the TREAT study in 2014. Demographic and clinical data, including sickle cell genotype, prior/alternative therapy, SCA-related complications, HU dosing, and laboratory values were abstracted from each patient's electronic medical record (EMR). Patients were identified using the EMR's sickle cell registry.
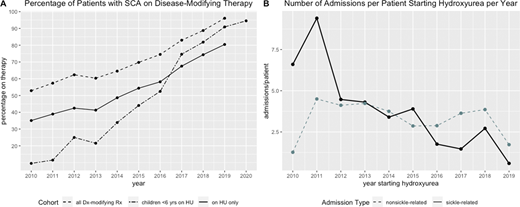
Results: We identified 439 patients with sickle cell disease followed at CCHMC from 2010-2019 (47% female, age range: 0-22 years); 275 had SCA (HbSS, HbS-0-thalassemia, or HbSD). The proportion of patients with SCA prescribed HU increased from 2010-19, from 35% in 2010 to 80% in 2019 with significantly more patients initiating HU during 2014-19 versus 2010-13 (average 20 versus 12 patients/yr, p = 0.0028, Figure 1A). The age of HU initiation was significantly lower during 2014-19 compared to 2010-13 (median = 2 y vs 6 y, p = 0.00028). Of 35 patients with SCA not on HU in 2019, 28 received chronic transfusions and the remaining 7 received no disease-modifying therapy with 3/7 patients not yet at the age to start HU. Ninety-six percent (53/55) of children with SCA born during 2014-19 were on treatment, including 52 on HU (median starting age = 10 months) and 1 on chronic transfusions; 45/52 (87%) were enrolled on TREAT or HOPS. With increased HU utilization during this study period, the number of admissions for sickle-related events was significantly lower in the 2014-19 group versus 2010-13 (2.8 vs 6.9 admissions/pt, p = 9.0 x 10-10) with no change in non-SCA related admissions, most commonly for fever (3.8 vs 4.0 admissions/pt, p = 0.8, Figure 1B).
Conclusions: HU has become the standard of care for children with SCA, beginning at 6-9 months of age, prior to the onset of acute and chronic complications. Despite widespread concerns that HU will not be accepted by patients and national trends demonstrating low rates of utilization, we have shown that a deliberate, systematic, and preventive approach to HU is possible and results in nearly universal acceptance of HU for young patients with SCA. This has translated to excellent laboratory responses and significantly fewer SCA-related clinical complications in our population. Our approach and improved patient outcomes can serve as a model for other programs to expand their HU treatment for more children with SCA.
Kalfa:Agios Pharmaceuticals, Inc: Consultancy, Research Funding; Forma Therapeutics, Inc: Research Funding. Malik:Aruvant Sciences, CSL Behring: Patents & Royalties; Aruvant Sciences, Forma Therapeutics, Inc.: Consultancy.

This feature is available to Subscribers Only
Sign In or Create an Account Close Modal