

Key Points
Kindlin-3 is recruited to the plasma membrane during rolling, before the induction of the high-affinity β2-integrin conformation.
Only a small fraction of kindlin-3 colocalizes with high-affinity integrins, and the PH domain is necessary for recruitment.

Abstract
Integrin-mediated neutrophil adhesion starts by arrest from rolling. Activation of integrins involves conformational changes from an inactive, bent conformation to an extended conformation (E+) with high affinity for ligand binding (H+). The cytoplasmic protein kindlin-3 is necessary for leukocyte adhesion; mutations of kindlin-3 cause leukocyte adhesion deficiency type 3. Kindlin-3 binds the β2-integrin cytoplasmic tail at a site distinct from talin-1, but the molecular mechanism by which kindlin-3 activates β2-integrins is unknown. In this study, we measured the spatiotemporal dynamics of kindlin-3 and β2-integrin conformation changes during neutrophil and HL-60 cell rolling and arrest under flow. Using high-resolution quantitative dynamic footprinting microscopy and kindlin-3–fluorescent protein (FP) fusion proteins, we found that kindlin-3 was recruited to the plasma membrane in response to interleukin-8 (IL-8) before induction of the H+ β2-integrin conformation. Intravital imaging revealed that EGFP-kindlin-3–reconstituted, kindlin-3–knockout neutrophils arrest in vivo in response to CXCL1. EGFP-kindlin-3 in primary mouse neutrophils was also recruited to the plasma membrane before arrest. Upon arrest, we found small clusters of high-affinity β2-integrin molecules within large areas of membrane-proximal kindlin-3 FP. Deletion of kindlin-3 or its pleckstrin homology (PH) domain in neutrophil-like HL-60 cells completely abolished H+ β2-integrin induction. IL-8 also triggered recruitment of the isolated kindlin-3 PH domain to the plasma membrane before arrest. In summary, we showed that the kindlin-3 PH domain is necessary for recruitment to the plasma membrane, where full-length kindlin-3 is indispensable for the induction of high-affinity β2-integrin.
Introduction
Neutrophils can reach virtually any tissue in the body, where they adhere to inflamed vascular endothelial cells, followed by transendothelial migration.1,2 In many tissues, adhesion is preceded by selectin-mediated rolling. Neutrophil adhesion occurs when a handful of clusters of β2-integrins acquire an extended (E+) high-affinity (H+) conformation that can bind integrin ligands such as intercellular adhesion molecules (ICAMs) to endothelial cells and stop the rolling neutrophil. This process is called arrest and precedes neutrophil spreading and crawling.
Neutrophil arrest is triggered by rapid β2-integrin activation initiated by ligation of chemokine receptors such as CXCR2. Neutrophils mainly express 2 β2-integrins: αLβ2, also known as LFA-1 or CD11a/CD18, and αMβ2, also known as Mac-1 or CD11b/CD18. Neutrophil3-5 and HL-60 cell6 arrest has been reconstituted in microfluidic devices coated with P-selectin and ICAM-1 and perfused with physiologic buffers at known wall shear stress.7 Arrest is triggered by interleukin-8 (IL-8), the main ligand for CXCR2, which is naturally expressed on human neutrophils and was stably transfected into our HL-60 cell line.8 In mouse neutrophils, CXCL1 triggers naturally expressed CXCR2, leading to neutrophil arrest.
Although all integrins can undergo activation, the degree of activation of integrins on blood cells is the most apparent. β2-Integrins are important adhesion and signaling molecules that are exclusively expressed on leukocytes. For β2-integrins, a 10 000-fold increase in affinity for ligand has been reported.9 Human β2-integrins express 2 activation epitopes that are recognized by 2 monoclonal antibodies, KIM127 and mAb24.10,11 The KIM127 epitope is in the knee of β2 and is not accessible when the integrin is bent; thus, KIM127 binding reports extension (E+). Once bound, KIM127 stabilizes the E+ conformation.12 The mAb24 binds to an epitope in the human β2 I-like domain that is accessible when the β2 I-like domain is bound to the internal ligand provided by the ligated αI domain13 and thus reports the high-affinity (H+) conformation. E−H− β2-integrins have very low affinity for ligands, E−H+ β2-integrins bind small molecule ligands14 and neutrophil cell surface ligands such as ICAM-1 and -3 in cis.7,15 E+H− β2-integrins support slow rolling but not arrest, and fully activated E+H+ β2-integrins mediate arrest.7,15 Because the epitopes for KIM127 and mAb24 are not conserved in mouse β2-integrins and no similar antibodies against mouse β2-integrins are available, most of the work reported here was completed in human HL-60 cells that were differentiated toward neutrophils.8,16
Activation of β2-integrins requires kindlin-3, but the sequence of events is not known and the mechanism remains unclear. Kindlin-3 contains a FERM (4.1, ezrin, radixin, moesin) domain consisting of the F1, F2, and F3 subdomains, and an additional atypical F0 domain at the N terminus.17 Unlike other FERM proteins, a pleckstrin homology (PH) domain is inserted in the F2 domain of kindlin-3. PH domains bind charged membrane phospholipids. The kindlin-3 PH domain has a higher affinity for phosphatidylinositol (3,4,5)-trisphosphate (PI(3,4,5)P3) than for PI(4,5)P2.18,19 Missense or nonsense mutations in the FERMT3 gene encoding human kindlin-3 underlie leukocyte adhesion deficiency-3 syndrome (LAD-3), which is characterized by bleeding caused by defective β3-integrin activation in platelets and by recurrent infections associated with defective neutrophil recruitment.20-22 LAD-3 has been recreated in mice by gene targeting with homologous recombination, introducing null mutations in the Fermt3 locus.23
Although LAD-3 was discovered a decade ago, very little is known about how kindlin-3 works mechanistically. Kindlin-3 binds to β2- and β3-integrin cytoplasmic domains, and this binding regulates integrin functions in platelets and neutrophils. One hypothesis is that kindlin-3 helps cluster integrin molecules, supported by the observation that kindlins selectively increase the binding of multivalent ligands to recombinant integrin-αIIbβ3 in cells, but not to monomeric integrins in nanodiscs.24 Kindlin-3 binds to an NPxY/F motif (NPKF in human β2) that is 9 amino acid residues C-terminal to another NPxY/F motif (NPLF in human β2) that binds talin-1, another important protein in integrin activation.25,26 The kindlin-3 binding site is in the unstructured portion of the β2 cytoplasmic tail. Kindlin-3 and talin-1 binding do not influence each other: they are neither synergistic nor cross-inhibitory.27,28 A second hypothesis is that kindlin-3 may be necessary for extended E+ β2-integrin to attain the high-affinity H+ conformation. This is supported by the observation that kindlin-3–deficient neutrophils roll normally, but completely fail to arrest.29 Talin-1–deficient neutrophils show significantly faster rolling and also fail to arrest. The β2-integrin–dependent reduction in rolling velocity is known to be dependent on E+H− β2-integrin molecules.30 Deletion of talin-1 in HL-60 cells prevented the induction of both E+ and H+ conformations and cell arrest, a phenotype similar to β2-knockout cells.31 The findings in these studies support the hypothesis that the absence of talin-1 prevents both E+ and H+ and the absence of kindlin-3 prevents the H+ conformation only.29,30
We recently developed quantitative dynamic footprinting (qDF),5,32 a total internal reflective fluorescence (TIRF)–based method suitable for live cell imaging that was later modified for simultaneous acquisition of 2 or 3 fluorochromes,7,33 by using homogeneous binding assays.34 qDF provides ∼10-nm spatial resolution in the vertical z-axis and ∼250-nm spatial resolution in the x- and y-axes with ∼1 frame per second time resolution in live cells.35 When variable-angle TIRF is used, the z-axis can be calibrated and the signal converted to a topographical map showing hills (microvilli) and valleys (areas between the microvilli).5 In combination with a β2-integrin conformation reporter antibody-binding assay in a flow chamber, qDF allows for examination of β2-integrin activation states in live cells under shear. Exposure of neutrophils to the chemokine IL-8 induces mAb24 binding (H+ conformation).7
The lack of imaging studies using fluorescence-labeled kindlins has precluded understanding the sequence of kindlin binding relative to integrin activation. In nanodiscs reconstituted with single integrin molecules, adding kindlin had no effect on the exposure of an integrin activation epitope.24 Yet, kindlin is strictly essential for integrin-mediated adhesion of cells.23,29,36 In Chinese hamster ovary (CHO) cells migrating on 2-dimensional fibronectin substrates, kindlin enters nascent focal adhesions in lockstep with integrins and before talin.37 Unlike CHO cells, leukocytes do not form focal adhesions.
The present study was designed to examine the temporal and spatial relationship between kindlin-3 and the activation of β2-integrin during chemokine-induced leukocyte arrest from rolling, and the role of the kindlin-3 PH domain.
Materials and methods
The conformation-specific antibodies mAb2438 and KIM12739 were conjugated to AlexaFluor 488 or 647 and DyLight 550 (Thermo Fisher Scientific), respectively. C57BL/6J mice (6-8 weeks old) were purchased from The Jackson Laboratory. Kindlin-3–deficient (Fermt3−/−) mice have been described.36 The plasmid encoding full-length human kindlin-3 protein was a gift from Nancy Hogg.18 The kindlin-3 coding sequence was tagged with EGFP or TagRFP and cloned into the vector pLVX-Het1 (Clontech) for lentiviral delivery or the pMSCV vector (pMIG; Addgene) for retroviral delivery. Kindlin-3 knockout (K3KO) HL-60 cells were generated with K3-single guide RNA GGGACTACATCGACTCGTCA. HL-60 cells were predifferentiated for 5 to 7 days in the presence of 1.3% dimethyl sulfoxide. Time-resolved antibody binding was recorded with an LSRII flow cytometer (BD). Microfluidic devices7,31 were coated with human or mouse P-selectin-Fc and ICAM-1-Fc and blocked with casein. Rolling and arrest were recorded by qDF5,33 on an IX71 inverted TIRF research microscope (Olympus) equipped with a 100× numerical aperture; 1.45 Plan-Apochromatic oil-immersion TIRF microscope objective; and 3-diode–pumped, solid-state lasers (CVI Melles Griot). Images were captured with a DV2 QuadView video coupler (Photometrics) and a 16-bit digital CCD camera (C10600-10B ORCA-R2; Hamamatsu). Three-dimensional (3D) reconstructions were generated by custom scripts in MatLab (MathWorks), as described previously.5,32 Fetal livers from E15.5-17.5 FERMT3−/− embryos were obtained by crossing Fermt3+/− heterozygous mice in timed pregnancies. Hematopoietic stem and progenitor cells (HSPCs) were isolated, expanded for 2 days, and infected with the viral supernatant of pMSCV-EGFP-K3. To reconstitute bone marrow, 8- to 12-week-old C57BL/6J mice were lethally irradiated and reconstituted by intravenous injection of 5 × 106 virally transduced HSPCs. The mouse cremaster muscle was prepared for intravital imaging40-42 with a 25×/0.95 numerical aperture water-immersion objective on a SP8 confocal microscope (Leica Microsystems). After 1 minute of baseline recording, 300 ng murine CXCL1 was injected through the carotid cannula. For western blot analysis, cells were lysed in Laemmli buffer (Bio-Rad) under reducing conditions. Statistical testing and graph creation were performed with Prism software (GraphPad). Data are expressed as means ± standard error of the mean (SEM). For comparisons between 2 groups, the 2-tailed Student t test was performed. For comparisons between more than 2 groups, 1-way ANOVA and the Tukey post hoc test were used. Differences reaching P < .05 were considered significant. Further details are given in the expanded supplemental Methods (available on the Blood Web site).
Results
To assess the effect of kindlin-3 on β2-integrin activation states, we first generated several stable cell lines based on CXCR2-expressing HL-60 cells.8 The CXCR2-expressing HL-60 cell line is a good model for chemokine-induced cell arrest and the arrest function is not affected by the CXCR2 overexpression (supplemental Figure 1). In preliminary experiments, we transfected kindlin-3 fusion proteins into wild-type (WT) HL-60 cells. The increase in membrane-proximal kindlin-3 induced by IL-8 was minimal, most likely because the unlabeled endogenous kindlin-3 was sufficient to activate β2-integrins and trigger arrest (supplemental Figure 2). To remove the endogenous kindlin-3 and to avoid the possible effects of overexpression, all experiments were conducted in HL-60 cells lacking FERMT3 (K3KO; supplemental Table 1; supplemental Figure 3). The deletion of kindlin-3 was confirmed at the protein level by western blot analysis (Figure 1A). Other cell lines included β2- and talin-1–deficient HL-60 cells.31

Kindlin-3 is essential for high-affinity integrin activation and HL-60 cell arrest. (A) A clustered regularly interspaced short palindromic repeats (CRISPR)/CRISPR-associated (Cas)–based gene-editing tool was used to delete kindlin-3 from HL-60 cells. Western blot detection of kindlin-3 was performed in WT or K3KO HL-60 cells. (B) Differentiated HL-60 cells deficient in kindlin-3 or integrin β2 (β2KO) were incubated with 100 ng/mL IL-8 in the presence of the fluorescently labeled, conformation-specific reporters mAb24 and KIM127. Real-time flow cytometry (left) was used to measure the binding of mAb24 and KIM127. Data are the change in median fluorescence intensity (MFI; right). Means ± SEM (n = 5, 5, and 3 individual experiments in WT, K3KO, and β2KO cells, respectively). (C) Fluorescent images of K3KO HL-60 cells expressing kindlin-3 fusion proteins (EGFP-K3 or TagRFP-K3). EGFP-K3 and TagRFP-K3 HL-60 were excited at 488 nm (top) and 561 nm (bottom), respectively. Scale bars, 10 µm. (D) WT, K3KO, or EGFP-K3 HL-60 cells rolled on P-selectin-Fc/ICAM-1-Fc substrate and were perfused with IL-8 (100 ng/mL) in a microfluidics-based cell arrest assay. Arrested cells per field were counted. (E) Integrin activation was measured by flow cytometry in WT and K3KO-, K3-EGFP–, and EGFP-K3–reconstituted HL-60 cells (n = 3, 3, 4, and 5, respectively). **P < .01; ***P < .001; ****P < .0001; n.s., not significant.
Kindlin-3 is essential for high-affinity integrin activation and HL-60 cell arrest. (A) A clustered regularly interspaced short palindromic repeats (CRISPR)/CRISPR-associated (Cas)–based gene-editing tool was used to delete kindlin-3 from HL-60 cells. Western blot detection of kindlin-3 was performed in WT or K3KO HL-60 cells. (B) Differentiated HL-60 cells deficient in kindlin-3 or integrin β2 (β2KO) were incubated with 100 ng/mL IL-8 in the presence of the fluorescently labeled, conformation-specific reporters mAb24 and KIM127. Real-time flow cytometry (left) was used to measure the binding of mAb24 and KIM127. Data are the change in median fluorescence intensity (MFI; right). Means ± SEM (n = 5, 5, and 3 individual experiments in WT, K3KO, and β2KO cells, respectively). (C) Fluorescent images of K3KO HL-60 cells expressing kindlin-3 fusion proteins (EGFP-K3 or TagRFP-K3). EGFP-K3 and TagRFP-K3 HL-60 were excited at 488 nm (top) and 561 nm (bottom), respectively. Scale bars, 10 µm. (D) WT, K3KO, or EGFP-K3 HL-60 cells rolled on P-selectin-Fc/ICAM-1-Fc substrate and were perfused with IL-8 (100 ng/mL) in a microfluidics-based cell arrest assay. Arrested cells per field were counted. (E) Integrin activation was measured by flow cytometry in WT and K3KO-, K3-EGFP–, and EGFP-K3–reconstituted HL-60 cells (n = 3, 3, 4, and 5, respectively). **P < .01; ***P < .001; ****P < .0001; n.s., not significant.
The expression level of β2-integrin in K3KO HL-60 cells was similar to that in WT cells (supplemental Figure 4). The absence of kindlin-3 in neutrophil-differentiated HL-60 cells resulted in a complete loss of induction of the mAb24 epitope in response to IL-8, as detected in time-resolved flow cytometry. This defect was complete, down to the level in β2-integrin–knockout cells31 and was indistinguishable from IgG control binding (supplemental Figure 5). Binding of KIM127 was also impaired but not eliminated (Figure 1B). These findings show that kindlin-3–deficient cells have a complete defect in acquiring high affinity (no H+) and a partial defect in acquiring β2-integrin extension (less E+).
We reasoned that high-resolution imaging could shed light on the function of kindlin-3. To use qDF, we transfected kindlin-3 fusion proteins with the fluorescent reporter protein EGFP or TagRFP (Figure 1C). These constructs (EGFP-K3 and TagRFP-K3) were expressed in K3KO HL-60 cells. The EGFP- or TagRFP-tagged kindlin-3 expression level was similar to that in the WT cells (supplemental Figure 6). As expected, K3KO cells showed no arrest in response to IL-8. The arrest defect was completely rescued by expression of EGFP-K3 (Figure 1D). Consistent with this, both the high-affinity H+ defect and the extension E+ defect were completely restored as measured by flow cytometry. Note that the C-terminal EGFP construct (K3-EGFP) did not fully restore kindlin-3 function (Figure 1E). Thus, all subsequent experiments were conducted by using N-terminal fusion.
Next, we labeled neutrophil-differentiated TagRFP-K3 HL-60 cells with the membrane label CellMask Deep Red (CMDR) and added Alexa Fluor 488–conjugated mAb24 to the perfusion medium in a microfluidic flow chamber. Smart segmentation image processing was used to remove background and generate binary masks of integrin clusters from raw images, preserving the cluster morphology and intensity with a better signal-to-noise ratio (supplemental Figure 7A-C). The membrane label clearly outlined the microvilli that were preserved during rolling (Figure 2A, first column; supplemental Movie 1) and was used for normalizing the intensities of kindlin-3 fusion proteins and antibodies.
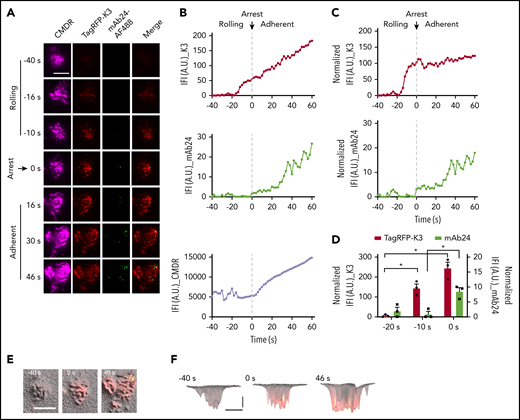
Kindlin-3 recruitment to the plasma membrane precedes integrin high affinity during arrest. (A) K3KO HL-60 cells expressing TagRFP-K3 were labeled with the membrane dye CMDR and rolled at 6 dyn/cm2 on coverslips coated with 5 μg/mL P-selectin-Fc and 10 μg/mL ICAM-1-Fc. Then, 100 ng/mL IL-8 was perfused in the presence of 1 μg/mL mAb24-Alexa Fluor 488 (mAb24-AF488). The cell rolling and arrest were observed using qDF imaging. Flow direction was from top to bottom. Merged images show mAb24-AF488 (green) and TagRFP-K3 (red). Scale bar, 5 μm. (B) Integrated fluorescence intensity (IFI, mean fluorescence intensity × area) of kindlin-3 and mAb24-AF488 antibody labeling is depicted as a function of time before and after arrest at 0 seconds. (C) IFI signal after normalization to the membrane (IFI_K3 or mAb24/IFI_CMDR×10 000). (D) Quantification of TagRFP-K3 and mAb24-AF488 binding during cell rolling and arrest in 3 independent experiments. (E-F) 3D distributions and dynamics of high-affinity β2 and kindlin-3 clusters in neutrophils. Membrane signals were converted to hills (microvilli) and valleys (space between microvilli). Top (E) and side (F) views of the 3D topography of membrane (gray) were overlaid with kindlin-3 (red) and mAb24 (green) clusters. Yellow, colocalization. (F) Horizontal bar represents 5 μm; vertical bar, 100 nm. *P < .05.
Kindlin-3 recruitment to the plasma membrane precedes integrin high affinity during arrest. (A) K3KO HL-60 cells expressing TagRFP-K3 were labeled with the membrane dye CMDR and rolled at 6 dyn/cm2 on coverslips coated with 5 μg/mL P-selectin-Fc and 10 μg/mL ICAM-1-Fc. Then, 100 ng/mL IL-8 was perfused in the presence of 1 μg/mL mAb24-Alexa Fluor 488 (mAb24-AF488). The cell rolling and arrest were observed using qDF imaging. Flow direction was from top to bottom. Merged images show mAb24-AF488 (green) and TagRFP-K3 (red). Scale bar, 5 μm. (B) Integrated fluorescence intensity (IFI, mean fluorescence intensity × area) of kindlin-3 and mAb24-AF488 antibody labeling is depicted as a function of time before and after arrest at 0 seconds. (C) IFI signal after normalization to the membrane (IFI_K3 or mAb24/IFI_CMDR×10 000). (D) Quantification of TagRFP-K3 and mAb24-AF488 binding during cell rolling and arrest in 3 independent experiments. (E-F) 3D distributions and dynamics of high-affinity β2 and kindlin-3 clusters in neutrophils. Membrane signals were converted to hills (microvilli) and valleys (space between microvilli). Top (E) and side (F) views of the 3D topography of membrane (gray) were overlaid with kindlin-3 (red) and mAb24 (green) clusters. Yellow, colocalization. (F) Horizontal bar represents 5 μm; vertical bar, 100 nm. *P < .05.
TagRFP-K3 was not visible at −40 seconds, where 0 indicates the moment of cell arrest. Kindlin-3 entered the TIRF field (∼150 nm into the cell footprint) at −20 seconds and continued to increase in a linear fashion before, during, and after arrest (Figure 2A, second column, and 2B, top panel). mAb24 binding did not appear until the time of arrest and then continued to increase after arrest (Figure 2A, third column, and 2B, middle panel). Superimposing the kindlin-3 on the mAb24 signal clearly showed that kindlin-3 recruitment to the area close to the plasma membrane (within 150 nm or closer) occurred before mAb24 binding. The microvilli started to flatten out after arrest (Figure 2A, first column, and 2B, bottom panel). To account for this effect, the kindlin-3 and antibody signals were normalized to the CMDR membrane signal (Figure 2C). Averaging data from 3 experiments showed a highly significant increase in kindlin-3 signal 10 seconds before arrest, but mAb24 signal did not appear until the time of arrest (Figure 2D; t = 0), which suggests that kindlin-3 must reside in the membrane-proximal compartment before β2-integrins can acquire the high-affinity conformation. Dye switch experiments confirmed this finding with EGFP-K3 and mAb24-DyLight 550 (supplemental Figure 8).
To formally address the relationship of microvilli with kindlin-3 recruitment and high-affinity β2-integrins, we used the membrane label (Figure 2A) to generate 3D topographical maps of the cell footprint, which is the area at the “bottom” of the cell, within 150 nm of the coverglass of the microfluidic chamber (Figure 2E-F). Superimposing high-affinity H+ β2-integrin as reported by mAb24 binding revealed small clusters of H+ integrin in areas that also showed signal for TagRFP-K3. Most of these H+ integrin clusters were located on the tips of microvilli. Kindlin-3 in the plasma membrane-proximal zone was much more widely expressed than mAb24 signal. Thus, not all areas with membrane-proximal kindlin-3 expressed the mAb24 epitope, suggesting that membrane-proximal kindlin-3 is required but is not sufficient for inducing the H+ integrin conformation.
Membrane-proximal kindlin-3 was arranged in 10 to 20 clusters in each rolling cell. The cluster size increased dramatically in the 10 seconds before arrest and continued to increase slightly after arrest (supplemental Tables 2 and 3). Together, the increased number and size of clusters resulted in a linear increase in the membrane-proximal kindlin-3 signal. The mAb24 signal showed fewer than 5 clusters in rolling cells, which increased at the time of arrest and continued to increase after arrest (supplemental Tables 2 and 3; supplemental Figure 7D-E). mAb24 clusters were ∼10 times smaller than kindlin-3 clusters; their size did not increase during rolling, but dramatically increased after arrest. Thus, the total mAb24+ area increased, starting at the time of arrest. We observed similar dynamics of kindlin-3 and mAb24 in dye switch experiments using EGFP-K3 and mAb24-DyLight 550 (supplemental Table 3). Differentiated HL-60 cells express β2- and β1-, not β3- or β7-integrins.43 We confirm that β1-integrins were expressed on HL-60 cells (supplemental Figure 9A). However, IL-8 stimulated β2-integrin activation, but not that of β1-integrins (supplemental Figure 9B-C).
Kindlin-3 contains a PH domain that is inserted in the F2 domain, splitting the F2 domain into F2a and F2b (supplemental Figure 3).18,19 To test the importance of the inserted PH domain in recruiting kindlin-3 to the membrane-proximal compartment, we expressed an EGFP-tagged kindlin-3 PH domain deletion (EGFP-K3delPH) mutant in K3KO cells (supplemental Table 1). The K3delPH mutant contained a deletion from amino acids 344 to 455 and thus lacked the entire PH domain. The expression levels of EGFP-K3 and EGFP-K3delPH were similar (Figure 3A). Western blot analysis (supplemental Figure 6) showed that EGFP-K3delPH ran at the expected size, suggesting that it folded properly. The K3delPH mutant completely failed to rescue mAb24 binding in response to IL-8. mAb 24 binding of HL-60 cells expressing mutant kindlin-3 was not significantly different from that of K3KO cells, whereas the WT kindlin-3 (positive control) completely rescued mAb24 binding (Figure 3B). The partial defect in KIM127 binding in K3KO cells was also completely rescued by WT kindlin-3, but not by the K3delPH mutant (Figure 3C). Unlike WT K3, the K3delPH mutant was did not restore arrest in K3KO cells under flow (Figure 3D). qDF imaging of EGFP-K3 or -K3delPH showed that EGFP-K3 but not EGFP-K3delPH was recruited to the membrane after IL-8 stimulation (Figure 3E-F). These data show that the PH domain is crucial for kindlin-3 recruitment.
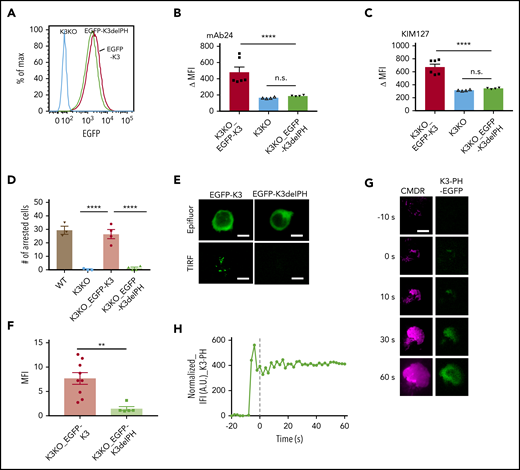
The PH domain of kindlin-3 is crucial for integrin activation and neutrophil adhesion. (A) K3KO HL-60 cells were reconstituted with WT kindlin-3 (K3KO_EGFP-K3) or K3 with PH domain truncation (K3KO_EGFP-K3delPH). The cells were sorted to match similar expression levels of EGFP. Flow cytometry with mAb24 and KIM127 antibodies was used to evaluate the IL-8–induced high-affinity integrin activation (B) and extension (C). The cell arrest assay (D) was performed on a substrate of P-selectin/ICAM-1 at a wall shear stress of 6 dyn/cm2. (E) Epifluorescence (Epifluor.) or TIRF image of EGFP-K3 and EGFP-K3dPH cells after stimulation with IL-8. (F) Quantification of the MFI of EGFP-K3 and EGFP-K3dPH recruited to the membrane. Means ± SEM of 9 EGFP-K3– and 5 EGFP-K3dPH–expressing cells. (G) qDF imaging of HL-60 cells expressing kindlin-3 PH domain-EGFP (green, right), kindlin-3-TagRFP (not shown) and labeled with membrane dye CMDR (magenta, left). Bar represents 5 μm. (H) The EGFP intensity was normalized to the membrane intensity, as in Figure 2. **P < .01; ****P < .0001.
The PH domain of kindlin-3 is crucial for integrin activation and neutrophil adhesion. (A) K3KO HL-60 cells were reconstituted with WT kindlin-3 (K3KO_EGFP-K3) or K3 with PH domain truncation (K3KO_EGFP-K3delPH). The cells were sorted to match similar expression levels of EGFP. Flow cytometry with mAb24 and KIM127 antibodies was used to evaluate the IL-8–induced high-affinity integrin activation (B) and extension (C). The cell arrest assay (D) was performed on a substrate of P-selectin/ICAM-1 at a wall shear stress of 6 dyn/cm2. (E) Epifluorescence (Epifluor.) or TIRF image of EGFP-K3 and EGFP-K3dPH cells after stimulation with IL-8. (F) Quantification of the MFI of EGFP-K3 and EGFP-K3dPH recruited to the membrane. Means ± SEM of 9 EGFP-K3– and 5 EGFP-K3dPH–expressing cells. (G) qDF imaging of HL-60 cells expressing kindlin-3 PH domain-EGFP (green, right), kindlin-3-TagRFP (not shown) and labeled with membrane dye CMDR (magenta, left). Bar represents 5 μm. (H) The EGFP intensity was normalized to the membrane intensity, as in Figure 2. **P < .01; ****P < .0001.
To test whether the kindlin-3 PH domain may be sufficient for targeting to the plasma membrane, we expressed and monitored the EGFP-tagged PH domain in HL-60 cells. Upon exposure to IL-8, the tagged PH domain appeared in the transfected with kindlin-3-TagRFP (TIRF) field, indicating that it went to the plasma membrane in response to IL-8 (Figure 3G-H).
Next, we retrovirally transduced K3KO mouse HSPCs with kindlin-3 and EGFP fusion proteins (EGFP-K3) and reconstituted lethally irradiated mouse recipients (Figure 4A). Similar to neutrophil-differentiated HL-60 cells, primary mouse neutrophils showed recruitment of kindlin-3 to the membrane-proximal compartment before arrest that continued after arrest (Figure 4B). Thus, kindlin-3 in primary mouse neutrophils mirrors the behavior of kindlin-3 in neutrophil-differentiated HL-60 cells during rolling and arrest. To address the in vivo relevance of kindlin-3 recruitment in neutrophil arrest, we performed intravital microscopy experiments in the well-established mouse cremaster model37,38 (Figure 4C). The neutrophils derived from nontransduced K3KO HSPCs (Figure 4C; purple) were unable to arrest, as expected, whereas all EGFP-K3 transduced neutrophils (Figure 4C; white) arrested in response to 300 ng CXCL-1 (Figure 4D).
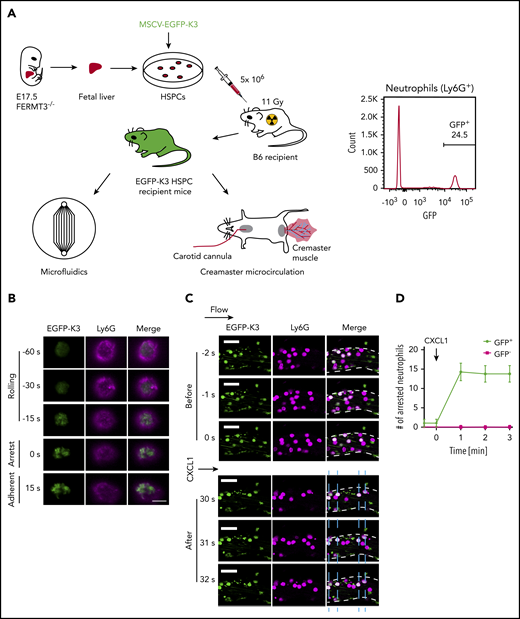
Recruitment of kindlin-3 to the plasma membrane during mouse neutrophil arrest. (A) Generation of mice reconstituted with EGFP-K3 and K3KO leukocytes for qDF and intravital imaging. Representative flow cytometric analysis of EGFP-K3 expression (percentage of positive cells indicated) in neutrophils from mouse whole blood (right). (B) Bone marrow cells were flushed from recipient mice reconstituted with EGFP-K3 (green) and labeled with 1 μg/mL anti-Ly6G antibody (Ly6G-AF647, magenta). qDF imaging was performed in mouse neutrophils rolling on mouse P-selectin/ICAM-1 substrate at 6 dyn/cm2 in a microfluidics-based flow chamber. Neutrophil arrest was induced by application of 10 ng/mL CXCL1. Flow direction was from top to bottom. Scale bar, 5 μm. Results are representative of 3 independent experiments. (C) Fluorescence (EGFP-K3 and Ly6G-AF647) images of neutrophils at the indicated time points before and after application of CXCL1. Vessel walls are outlined by broken white lines. Horizontal arrow indicates the direction of blood flow. Blue dashed lines indicate positions of arrested neutrophils (white, EGFP-K3+Ly6G+). Scale bars, 20 μm. (D) The number of adherent neutrophils in mouse cremaster muscle venules of reconstituted mice, before and after injection of 300 ng CXCL1 via the carotid artery catheter. Data are shown as means ± SEM; n = 4 observations in 4 vessels after 4 individual CXCL1 injections. MSCV, murine stem cell virus.
Recruitment of kindlin-3 to the plasma membrane during mouse neutrophil arrest. (A) Generation of mice reconstituted with EGFP-K3 and K3KO leukocytes for qDF and intravital imaging. Representative flow cytometric analysis of EGFP-K3 expression (percentage of positive cells indicated) in neutrophils from mouse whole blood (right). (B) Bone marrow cells were flushed from recipient mice reconstituted with EGFP-K3 (green) and labeled with 1 μg/mL anti-Ly6G antibody (Ly6G-AF647, magenta). qDF imaging was performed in mouse neutrophils rolling on mouse P-selectin/ICAM-1 substrate at 6 dyn/cm2 in a microfluidics-based flow chamber. Neutrophil arrest was induced by application of 10 ng/mL CXCL1. Flow direction was from top to bottom. Scale bar, 5 μm. Results are representative of 3 independent experiments. (C) Fluorescence (EGFP-K3 and Ly6G-AF647) images of neutrophils at the indicated time points before and after application of CXCL1. Vessel walls are outlined by broken white lines. Horizontal arrow indicates the direction of blood flow. Blue dashed lines indicate positions of arrested neutrophils (white, EGFP-K3+Ly6G+). Scale bars, 20 μm. (D) The number of adherent neutrophils in mouse cremaster muscle venules of reconstituted mice, before and after injection of 300 ng CXCL1 via the carotid artery catheter. Data are shown as means ± SEM; n = 4 observations in 4 vessels after 4 individual CXCL1 injections. MSCV, murine stem cell virus.
Taken together, our results confirm that kindlin-3 is necessary for neutrophil arrest in vitro and in vivo. In our study, kindlin-3 was recruited to the plasma membrane starting at ∼10 to 20 seconds before arrest, and the presence of the kindlin-3 PH domain was necessary for this recruitment.
Discussion
Our data showed that kindlin-3 was recruited to the plasma membrane of rolling leukocytes before arrest. The lag time between kindlin-3 recruitment and the appearance of high-affinity β2-integrin as detected by the expression of the mAb24 epitope was ∼10 seconds. Kindlin-3 lacking the PH domain did not restore leukocyte arrest and mAb24 binding, suggesting that kindlin-3 must bind charged membrane phospholipids such as PI(3,4,5)P3 on the inner leaflet of the plasma membrane.18,19 When IL-8 ligates CXCR2, Gα dissociates from Gβγ, which activates phosphoinositide-3 kinase (PI3K)-γ and -δ. These enzymes phosphorylate PI(4,5)P2 to PI(3,4,5)P3.44,45 Our data suggest that this transition recruits kindlin-3 to the plasma membrane, where it can interact with both PI(3,4,5)P3 and the β2-integrin tail. Our data show that the interaction of kindlin-3 with the charged plasma membrane phospholipids depends on the PH domain, because kindlin-3 without the PH domain fails to rescue the arrest function and the induction of the mAb24 epitope. Conversely, PH domain alone is recruited to the membrane-proximal compartment.
Kindlin-3 in the plasma membrane-proximal zone was much more widely distributed than mAb24 signal. The larger area of kindlin-3 recruited to the membrane and the propensity of the isolated PH domain to be recruited indicates that the K3 recruitment mainly depends on the known conversion of PI(4,5)P2 to PI(3,4,5)P3. It further suggests that kindlin-3 alone is necessary but not sufficient for the high-affinity conformation. Indeed, minimal amounts of kindlin-3 suffice for basal platelet and leukocyte functions in mice.46 Thus, there is more kindlin-3 than is needed for β2-integrin activation. It is possible that kindlin-3 binds β1-integrins, which are known to be expressed in HL-60 cells.43 However, β1-integrins are not activated in response to IL-8. Our experimental flow chamber system contained no β1-integrin ligand; thus, β1-integrins were not involved in arrest under the conditions studied.
Kindlin-3 knockout completely abolished the integrin H+ conformation, and partially impaired induction of the E+ conformation in response to IL-8. A previous study showed that mAb24 binding were abolished by kindlin-3 knockdown in undifferentiated HL-60 cells.29 In that study,29 constitutively active Rap1 protein was used. It bypasses the chemokine signaling, including the formation of PI(3,4,5)P3, which provides a likely explanation for the different results. The β2-integrin E+ conformation is already present during rolling without IL-8 stimulation (supplemental Movie 2). The partial defect in E+ induction in K3KO cells, but unaltered rolling suggests that the remaining induction of KIM127 reactivity was sufficient to maintain normal rolling.
We know from previous work that not all mAb24+ integrin molecules are fully active (extended and high affinity).7 The mAb24 signal detected in this study very likely represents both high-affinity–bent (E-H+) and high-affinity–extended (E+H+) β2-integrins. Interestingly, β2-integrins are unable to assume the high-affinity (mAb24+) conformation in the absence of kindlin-3, or when kindlin-3 lacks the PH domain. Exactly why this is the case is still unknown. The main defect seems to be a complete loss of mAb24 epitope and a partial reduction of extension as reported by the mAb KIM127. From previous work,25,26,29,47 it is known that talin-1 is required for integrin activation. Our current data provide evidence that talin-1 is not sufficient. It is known that kindlin-3 and talin-1 binding to integrin are not mutually exclusive.26,27 Most likely, both kindlin-3 and talin-1 are bound to active integrin simultaneously.
The phenotype of kindlin-3 deficient β2-integrins is different from the phenotype observed in β2-integrins, in which a kink mutation was inserted in the β2 transmembrane domain.31 The β2 kink mutation causes a partial defect in KIM127 binding with no defect in mAb24 binding. The absence of kindlin-3 causes a similar partial defect in KIM127 binding but completely abolishes mAb24 binding. Thus, the high-affinity β2-integrin conformation requires kindlin-3, but not a stiff β2 transmembrane domain.
The finding that kindlin-3 recruitment precedes integrin activation is consistent with previous findings in CHO cells,37 where integrins assemble into focal adhesions under static conditions and at a much slower pace than in rolling neutrophils. It is not known whether kindlin-3 can organize integrin clusters consisting of more than 2 integrin molecules. The size and shape of the clusters of high-affinity β2-integrin, as revealed by superresolution microscopy,15 suggest that elongated arrays of active β2-integrin molecules exist. We speculate that these may be multiple subunits of kindlin-348 bound to integrin β2 chains and aligned by other molecules. Candidates of such molecules indirectly mediating clustering include α-actinin, vinculin, and talin-1.37
Although our data clearly demonstrated that kindlin-3 was recruited to the plasma membrane through its PH domain before integrin activation, our findings still do not resolve the conundrum of the 2 forms of high-affinity β2-integrin: on activated neutrophils, approximately half of the mAb24+ (H+) integrin molecules are bent and thus cannot bind ligand in trans,7,15 and the other half are extended KIM127+mAb24+ (E+H+), can bind ICAM-1 in trans, and therefore can mediate cell arrest. Our data suggest that kindlin-3 is necessary for both E−H+ and E+H+ β2-integrin conformations. Neither conformation is detectable when kindlin-3 is absent. Together with previous findings showing that talin-1 is necessary for β2-integrin extension,29 this suggests a sequential model. First, chemokine binds the chemokine receptor; second, Gα dissociates from Gβγ; third, Gβγ activates PI3K-γ and -δ; fourth, the PI3 kinases produce PI(3,4,5)P3; fifth, kindlin-3 is recruited to PI(3,4,5)P3; and sixth, membrane-bound kindlin-3 orients the β2-integrin molecules relative to each other and enforces a fixed distance. Kindlin-3, in the presence of talin-1, allows for the full activation of β2-integrins to the E+H+ conformation. Previous in vivo data on talin-1 knockout and kindlin-3 knockout have shown that talin-1 is important for slow rolling and arrest, whereas kindlin-3 is important for arrest.29 These observations are compatible with a sequential model in which kindlin-3 binding precedes β2-integrin activation. Future work directly imaging kindlin-3 and talin-1 in the same cell at the same time is needed to determine when talin-1 binding occurs. Such imaging cannot be done using lentiviral technology, because talin-1 is too large to be packaged into a lentivirus. Our model is consistent with the experimentally observed stoichiometry between kindlin, talin, and integrin (1:1:1) in CHO cells.37
In summary, our results showed that kindlin-3 is recruited to the plasma membrane ∼10 seconds before β2-integrin activation in live rolling and arresting HL-60 cells and primary mouse neutrophils. This process requires the PH domain. Conversely, the isolated PH domain recruited to the plasma membrane in response to IL-8. Our study contributes to the mechanistic understanding of kindlin-3 function in integrin activation and how its absence causes the disease phenotype of LAD-3.
The data that support the findings of this study are available from the corresponding author upon reasonable request.
The online version of this article contains a data supplement.
The publication costs of this article were defrayed in part by page charge payment. Therefore, and solely to indicate this fact, this article is hereby marked “advertisement” in accordance with 18 USC section 1734.
Acknowledgments
The authors thank R. Fässler for providing the kindlin-3 knockout mouse, Ann Richmond for the HL-60 cell line, and Nancy Hogg for the kindlin-3 plasmid.
This work was supported by National Institutes of Health, National Heart Lung and Blood Institute grants HL078784 (K.L.) and R01HL145454 (Z.F.), Postdoctoral Fellowship 19POST34450228 (L.W.), and Career Development Award 18CDA34110426 (Z.F.) from the American Heart Association.
Authorship
Contribution: L.W. and K.L. designed experiments. L.W. performed most of the experiments and the data analyses; A.M. performed intravital imaging; P.R. performed western blot analysis; S.M. coded part of the custom scripts on MatLab and performed data analysis; Z.F. helped with the qDF setup; H.S. and M.H.G. provided essential reagents; A.R.G. conducted structural analysis; K.L. supervised the project; K.L. and L.W. wrote the manuscript; and all authors provided constructive suggestions to experimental design and read and revised the manuscript.
Conflict-of-interest disclosure: The authors declare no competing financial interests.
Correspondence: Klaus Ley, Laboratory of Inflammation Biology, La Jolla Institute for Immunology, 9420 Athena Circle Dr, La Jolla, CA 92037; e-mail: klaus@lji.org.

This feature is available to Subscribers Only
Sign In or Create an Account Close Modal