

Engineering a proper transgenic mouse model for the blood cancer multiple myeloma has been a bumpy road, with few successes and many disappointments. In this issue of Blood, 1 achieve an important milestone, providing a model that presents several features mimicking aggressive myeloma in humans.
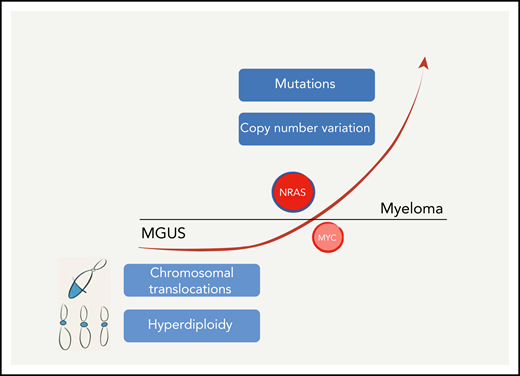
Wen et al have generated a mouse that models one of the leading genetic lesions present in myeloma, NRASQ61R, pushing MGUS toward a more aggressive stage of the disease. However, several other genetic lesions have so far eluded modeling. These alterations, central for the development of the disease, are present as early as in the MGUS phase, including hyperdiploidy and several hallmark chromosomal translocations, which deregulate the expression of various genes (cyclins, MMSET, MAF, and others), or appear in later stages of the disease, elicited by somatic mutations or copy number aberrations, affecting a large number of oncogenes and tumor-suppressor genes.
Wen et al have generated a mouse that models one of the leading genetic lesions present in myeloma, NRASQ61R, pushing MGUS toward a more aggressive stage of the disease. However, several other genetic lesions have so far eluded modeling. These alterations, central for the development of the disease, are present as early as in the MGUS phase, including hyperdiploidy and several hallmark chromosomal translocations, which deregulate the expression of various genes (cyclins, MMSET, MAF, and others), or appear in later stages of the disease, elicited by somatic mutations or copy number aberrations, affecting a large number of oncogenes and tumor-suppressor genes.
Mouse models remain critical tools for understanding the biology of a particular cancer by defining the relevance of specific genetic lesions in tumorigenesis and the interactions between cancer cells and their surrounding microenvironment, and for testing drugs in a setting that is closer to the human disease than tumor cell lines grown in a plate.2 In the case of myeloma, a cornucopia of increasingly sophisticated transplantation mouse models have been proposed, while the generation of reliable transgenic models has lagged behind.3 One of the reasons transgenic models have proven difficult to generate is the frustrating inability in identifying reliable promoters driving the expression of genes specific for the elusive myeloma cell of origin. As such, many efforts to generate a myeloma model using B lymphocyte–specific promoters to propel the expression of myeloma-pertinent oncogenes or elicit the ablation of context-relevant tumor-suppressor genes have resulted in models that instead develop lymphomas or aggressive extramedullary plasma cell overgrowths, ie, plasmacytomas.
One of the most successful attempts to overcome these limitations came a few years ago, when the Chesi and Bergsagel laboratory developed the Vκ*MYC model,4 engineered exploiting an ingenious molecular trick. The immunoglobulin sequences of B lymphocytes residing within the lymph node germinal centers undergo somatic hypermutation, whereby the variable diversity joining (VDJ) locus is modified in a handful of nucleotides to finely tune the antibody affinity for antigens. In the Vκ*MYC mouse, a stop codon has been inserted in the V-κ sequence, which is placed upstream of the human MYC oncogene. Somatic hypermutation reverts the stop codon in a small subset of germinal center B lymphocytes eliciting MYC expression, which in turn leads to an expansion of plasma cells and a phenotype in the mice reminiscent of human indolent myeloma.
Wen et al have layered on top of the Vκ*MYC model another powerful oncogene, RAS, which is frequently mutated in myeloma5,6 and is implicated in the crucial transition from myeloma premalignant condition, monoclonal gammopathy of undetermined significance (MGUS), into the full blown disease5 (see figure). However, at this point, a note concerning the RAS gene family is in order. The long-standing idea that all RAS proteins are created equal and behave similarly is not sustainable anymore. In fact, KRAS, HRAS, and NRAS, even when mutated, are bestowed with biological activities that are quite different among themselves. These differences are, and not in a subtle way, largely due to their C-terminal hypervariable region.7 Evidence for this comes from both the laboratory8 and the clinic,9 even shown in myeloma.10 There is indeed wide variability across cancers in which RAS family members are mutated, including variability in the percentage of cases and which residues are mutated.7,9 In myeloma, somatic mutations frequently occur in the KRAS and NRAS genes.6 Wen et al decided to exploit the conditional expression of endogenous NRASQ61R,9 a mutation rarely present in other hematological cancers, and which confers transcriptomic changes that are different from the ones present in KRAS myelomas.10
As a result, the mouse model presented by Wen et al, dubbed VQ, develops a disease that encompasses several of the key features that are present in aggressive myeloma patients. Besides the clinical manifestations that are present also in the Vκ*MYC mouse, in the VQ model plasma cells display a tumultuous growth in the bone marrow as well as in other tissues, including the spleen, lymph nodes, and the kidneys. Accordingly, VQ mice present a more profound impairment of hematopoiesis and ensuing anemia and increased immunoglobulin deposition in the kidneys, when compared with the Vκ*MYC model. Attesting to the aggressive nature of the disease in the VQ mouse, plasma cells are highly proliferative, and, intriguingly, present a gene expression pattern reminiscent of the high-risk myeloma signature (UAMS-70). As expected, the survival of the VQ mice is severely affected, with the animals living only for a few months.
The translational relevance of the VQ model has also been explored by Wen et al and is quite remarkable. For example, as it has been for the Vκ*MYC model,4 the VQ mouse was successfully tested as a drug screening in vivo platform, with drugs already used in the clinic to treat myeloma patients or more experimental compounds. In addition, VQ myeloma cells preserve both the PD1 and the TIGIT immune checkpoint pathways, which mediate T-cell suppression in myeloma patients. As such, the VQ mice could be exploited for the preclinical evaluation of immunotherapeutic strategies in an aggressive myeloma setting. Finally, tumor cells derived from this model could be transplanted into sublethally irradiated syngeneic recipients for several passages and, importantly, could be transduced with both retro- and lentiviruses, thus enabling additional genetic manipulations.
Thus, are we there yet? Is this the end of the journey? The model presented herein represents an important step forward, toward the generation of a reliable mouse model of aggressive myeloma. However, in solid tumors, the knowledge of the underlying genetic lesions has led to the generation of increasingly refined mouse models, as a result of the accrual of layers of genetic engineering.2 Although MYC and NRAS certainly represent crucial genetic events in the development of the disease,4,5 myeloma is a genetically heterogeneous disease, with several other genes and pathways that are engaged in its development, in specific patient subsets.5 These lesions have not yet been properly modeled. There is indeed the possibility that the platform described in this study may represent the foundation for models where additional lesions could be engineered, to capture more closely the puzzling intra- and interpatient heterogeneity of human myeloma.
REFERENCES
Conflict-of-interest disclosure: The author declares no competing financial interests.

This feature is available to Subscribers Only
Sign In or Create an Account Close Modal