

In this issue of Blood, demonstrate that the xenotropic and polytropic retrovirus receptor 1 (XPR1) serves as a phosphate transporter that controls platelet polyphosphate content and thereby modulates thrombosis.1
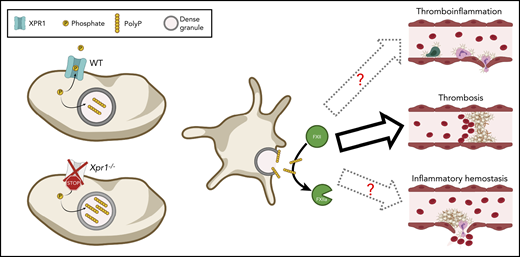
XPR1-mediated phosphate export reduces platelet polyP content and thereby limits platelet-dependent FXII activation and thrombosis. In the absence of functional XPR1, phosphates are no longer exported, which results in increased polyP levels in dense granules of platelets. Consequently, at sites of platelet activation, more polyP is released, which results in increased thrombosis as demonstrated by Mailer et al. It will be interesting to further investigate whether elevated polyP levels as a consequence of reduced XPR1 activity would also translate into increased thromboinflammation and/or might reduce inflammatory bleeding. This figure was created using BioRender.com.
XPR1-mediated phosphate export reduces platelet polyP content and thereby limits platelet-dependent FXII activation and thrombosis. In the absence of functional XPR1, phosphates are no longer exported, which results in increased polyP levels in dense granules of platelets. Consequently, at sites of platelet activation, more polyP is released, which results in increased thrombosis as demonstrated by Mailer et al. It will be interesting to further investigate whether elevated polyP levels as a consequence of reduced XPR1 activity would also translate into increased thromboinflammation and/or might reduce inflammatory bleeding. This figure was created using BioRender.com.
At sites of vascular injury, activated platelets release their granule content, which amplifies the activation response and promotes local activation of the coagulation cascade resulting in the formation of a fibrin-rich clot that seals the wound. This concerted action of platelets and the coagulation system has to be regulated with high fidelity to enable normal hemostasis while preventing uncontrolled thrombus growth, which may lead to thrombotic vessel occlusion, ischemia, and infarction of vital organs. In the classic cascade or waterfall models of blood coagulation,2 fibrin formation can be initiated through either of 2 converging cascades designated the extrinsic and intrinsic pathways. The essential function of the factor VIIa (FVIIa)/tissue factor complex–triggered extrinsic pathway is firmly established, and functional defects in these components severely compromise blood coagulation in vivo.2 In sharp contrast, hereditary deficiency of FXII (Hageman factor), the protease that triggers the intrinsic pathway after being activated by polyanionic surfaces (contact activation), is not associated with spontaneous hemorrhage or excessive injury-related bleeding, which led to the hypothesis that FXII is not required for fibrin formation in vivo. This picture changed in 2005, when Renné et al3 showed that FXII-deficient mice, although they display normal hemostasis, were profoundly protected from injury-induced occlusive thrombus formation in different thrombosis models. These findings provided the first experimental evidence that the mechanisms that drive thrombosis may in part be distinct from those required for hemostasis, which might allow pharmacologic prevention of thrombosis without increasing the bleeding risk. This was experimentally confirmed a few years later using the first specific FXII inhibitor, rHA-Infestin-4.4 In addition to the intrinsic coagulation pathway, FXII also triggers the proinflammatory kallikrein-kinin system (KKS) that liberates the vasoactive peptide hormone bradykinin.5 This dual function puts FXII in a central position at the interface between thrombotic and inflammatory pathways, which makes it a prime target to interfere with thrombo-inflammatory diseases such as ischemic stroke.6
The recognition of FXII as a key player in thrombotic and thromboinflammatory disease settings sparked intense interest in the mechanisms underlying its activation in vivo. Despite a long-recognized role of activated platelets in promoting FXII activation, it took until 2009 to identify platelet polyphosphate (polyP) as the underlying factor in this process.7 Polyphosphates are linear polymers of orthophosphate that are stored in platelet-dense granules and released upon activation. Following this seminal discovery, several groups have identified that platelet polyP contributes to procoagulant and proinflammatory pathways. Polyphosphates can also be secreted from microorganisms and some immune cells and, depending on the polyP chain length, have been shown to promote plasmatic coagulation, modulate inflammation by inhibiting the complement system, and trigger bradykinin release,5,8 and inhibition of polyP has been suggested as a novel antithrombotic and anti-inflammatory strategy.8,9 Although platelet polyP is shorter than bacterial polyP and is thus considered to be less potent, it has been shown to increase endothelial cell permeability, promote the formation of neutrophil extracellular traps, and modulate fibrin clot structure,5,8
Mailer et al have added another piece to the platelet polyP puzzle by identifying XPR1 as a major phosphate exporter in platelets. XPR1 is expressed in murine and human platelets, and pharmacologic inhibition of this transporter in heterologous cells or on human platelets resulted in the accumulation of intracellular polyP. Similar results were obtained in mice lacking XPR1, specifically in megakaryocytes and platelets (Xpr1fl/fl, Pf4-Cre), indicating that XPR1, by exporting excess phosphates, regulates homeostatic polyP levels in platelets. Of note, polyP levels were affected independently of their chain length because soluble and longer platelet polyP were affected equally. As a consequence of the elevated polyP content, the procoagulant activity of these platelets was increased, which translated into increased thrombus formation and fibrin deposition in vitro and in vivo. Furthermore, the authors identified FXII as the main target of platelet polyP, because FXII blockade abrogated the procoagulant effect of elevated platelet polyP levels. This FXII dependency is interesting, because platelet polyP has been proposed to be a weak FXII activator, and its procoagulant effects were ascribed to accelerated FXI activation and blockade of the tissue factor pathway inhibitor rather than to FXII activation.8
Future studies, using either Xpr1fl/fl, Pf4-Cre mice, which have higher polyP levels, or mice lacking the inositol hexakisphopshate kinase 1 (Ip6k1−/−), which have lower platelet polyP levels,10 may help shed light on the specific role of platelet polyP in different thrombo-inflammatory disease settings. Mice with defective dense granule secretions that are thus unable to release platelet polyP are protected from arterial thrombosis as well as thrombo-inflammatory cerebral infarct progression after experimental ischemic stroke.6 Although the antithrombotic protection is clearly a result of the abolished platelet adenosine 5′-diphosphate/adenosine triphosphate release, the reduced cerebral thromboinflammation may, at least in part, be a consequence of the abrogated polyP-induced FXII activation. FXII has been shown to facilitate various aspects of thromboinflammation, from modulating the KKS to fine-tuning neutrophil responses to neuroinflammation in the context of autoimmune encephalomyelitis.5 It remains to be seen whether the platelet polyP-FXII axis also triggers these reactions and how it affects the KKS.
Another question arising from the work of Mailer et al is whether inhibiting XPR1 and thereby enhancing the procoagulant potential of platelets might be a suitable strategy for limiting excessive spontaneous or injury-related bleeding. XPR1-deficient mice did not display shortened tail bleeding times; however, these mice were healthy. Under inflammatory conditions, maintenance of vascular integrity and hemostasis depend on mechanisms that are in part distinct from those required for normal hemostasis.6 It will be interesting to study the significance of platelet polyP and its regulation in models of thromboinflammation and inflammatory bleeding (see figure). Of note, mice with reduced platelet polyP levels had slightly prolonged tail bleeding times,10 indicating that platelet polyP can contribute to hemostasis, perhaps even in an FXII-independent manner.3,8,10 Because XPR1 inhibition would elevate platelet polyP levels, the procoagulant effect of such a treatment would be observed only at sites of vascular injury where platelets adhere and release their granule content, including polyP.
Conflict-of-interest disclosure: The authors declare no competing financial interests.

This feature is available to Subscribers Only
Sign In or Create an Account Close Modal