


TO THE EDITOR:
We describe here a family with a high incidence of B-cell precursor acute lymphoblastic leukemia (BCP-ALL) affecting 3 children (II.1, II.2, and II.3) at 11, 17, and 25 years old, respectively (Figure 1A,C; supplemental Figure 1, available on the Blood Web site). Both parents were asymptomatic. After a first relapse, the proband (II.2) underwent a familial allogeneic hematopoietic stem cell transplantation (HSCT) with his 11-year-old younger brother (II.3) leading to a complete remission (CR) for 20 years, but he relapsed a second time and died very soon after intracranial hemorrhage. His sister (II.1) developed BCP-ALL for which she was treated by chemotherapy leading to a CR even 30 years after her initial diagnosis. II.3 developed BCP-ALL 14 years after being an HSCT donor for II.2 and died of infectious complications after HSCT from an unrelated donor (supplemental Figure 1).
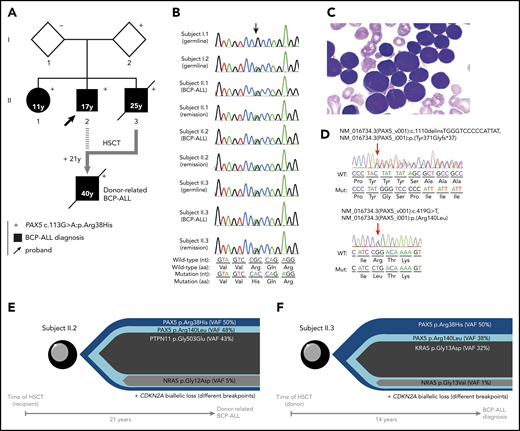
Familial BCP-ALL with heterozygous germline PAX5 R38H mutation. (A) The germline R38H variant was shown to be inherited from 1 parent (I.2) who is so far an asymptomatic carrier with no history of cancer at age 68 years. The proband (II.2, indicated by an arrow) developed BCP-ALL at age 17 years. He relapsed 2 years after his initial diagnosis and was allografted with his brother (II.3) as a donor 14 years before II.3 also developed BCP-ALL. II.2 relapsed at age 40 years and died soon thereafter as a result of an intracranial hemorrhage. His sister (II.1) developed BCP-ALL at age 11 years for which she received chemotherapy without HSCT. She is still in CR more than 30 years after her initial diagnosis. The proband’s younger brother (II.3) also developed also BCP-ALL at age 25 years and died of infectious complications after HSCT from an unrelated donor. (+) Indicates the presence of the R38H germline mutation. (B) Sanger sequencing of PAX5 mutation in samples of I.1, I.2, II.1, II.2, and II.3; the location of the mutation is indicated by an arrow demonstrating a germline origin. The nucleotide and protein sequences are indicated at the bottom of the panel. (C) Representative image of May-Grünwald-Giemsa–stained bone marrow smear at BCP-ALL diagnosis in individual II.1. (D) Somatic PAX5 mutations in leukemic samples from individuals II.1 (top) and II.3 (bottom). Positions of mutations are indicated by red arrows. (E-F) Leukemic architecture at BCP-ALL diagnosis in individual II.3 (donor) (E) and at donor-related BCP-ALL in individual II.2 (recipient) (F). Variant allele frequency (VAF) for each mutation is indicated.
Familial BCP-ALL with heterozygous germline PAX5 R38H mutation. (A) The germline R38H variant was shown to be inherited from 1 parent (I.2) who is so far an asymptomatic carrier with no history of cancer at age 68 years. The proband (II.2, indicated by an arrow) developed BCP-ALL at age 17 years. He relapsed 2 years after his initial diagnosis and was allografted with his brother (II.3) as a donor 14 years before II.3 also developed BCP-ALL. II.2 relapsed at age 40 years and died soon thereafter as a result of an intracranial hemorrhage. His sister (II.1) developed BCP-ALL at age 11 years for which she received chemotherapy without HSCT. She is still in CR more than 30 years after her initial diagnosis. The proband’s younger brother (II.3) also developed also BCP-ALL at age 25 years and died of infectious complications after HSCT from an unrelated donor. (+) Indicates the presence of the R38H germline mutation. (B) Sanger sequencing of PAX5 mutation in samples of I.1, I.2, II.1, II.2, and II.3; the location of the mutation is indicated by an arrow demonstrating a germline origin. The nucleotide and protein sequences are indicated at the bottom of the panel. (C) Representative image of May-Grünwald-Giemsa–stained bone marrow smear at BCP-ALL diagnosis in individual II.1. (D) Somatic PAX5 mutations in leukemic samples from individuals II.1 (top) and II.3 (bottom). Positions of mutations are indicated by red arrows. (E-F) Leukemic architecture at BCP-ALL diagnosis in individual II.3 (donor) (E) and at donor-related BCP-ALL in individual II.2 (recipient) (F). Variant allele frequency (VAF) for each mutation is indicated.
Because the family history was compatible with a germline transmission of the disease, whole-exome sequencing was performed on II.3, which identified a PAX5 germline heterozygous c.113G>A mutation resulting in an R38H substitution. The R38H substitution is predicted to alter the protein affinity to DNA1 and concerns a highly conserved residue located within the N-terminal DNA-binding paired domain (supplemental Figure 2A-C; supplemental Tables 1 and 2). This mutation is associated with leukemic progression in murine models with Pax5 haploinsufficiency2 or PAX5-ELN fusion.3 This mutation was subsequently detected in all affected patients at BCP-ALL diagnosis and at remission, and in 1 asymptomatic parent without a history of malignancy (Figure 1B; supplemental Figure 1; supplemental Table 3). PAX5 somatic alterations are present in one-third of the patients with sporadic BCP-ALL,4,5 but PAX5 germline mutations leading to BCP-ALL have been identified only recently.6-8 PAX5 encodes a critical transcription factor for B-cell differentiation9-11 repressing B-lineage “inappropriate” gene expression and promoting the transcription of specific B-cell genes.10-14
As described in other germline PAX5 pedigrees,6-8 the existence of asymptomatic carriers and the absence of immunodeficiency before the onset of BCP-ALL suggest the requirement of additional genetic alterations that lead to the development of leukemia. Patients II.2 and II.3 had a normal karyotype at the BCP-ALL stage. Additional cytogenetic abnormalities were found in II.2 at his first (before transplantation) and second (after transplantation with his brother, consequently named II.3/2) relapses. Somatic CDKN2A homozygous loss and RAS pathway mutations, recurrent features of BCP-ALL, were detected in all analyzed BCP-ALL samples (II.2, II.3, and II.3/2) (supplemental Figure 3; supplemental Tables 3 and 4). A second PAX5 mutation was detected at diagnosis: Y371fs in II.1 and R140L in II.3 and II.3/2 (Figure 1D-F; supplemental Table 3). Next-generation sequencing of II.3 complementary DNA (cDNA) showed that these 2 PAX5 mutations were mainly detected on different alleles (data not shown).
More than 20 years separate HSCT allograft in II.2 from his donor-related (II.3/2) leukemia and 14 years from the allograft to diagnosis of BCP-ALL in II.3 (supplemental Figure 1). The leukemic samples from II.2 and II.3 harbored distinct IGH clonal rearrangements (supplemental Figure 4), suggesting that the R140L mutation occurred independently in both patients. Interestingly, the R140L mutation is recurrently associated with the R38H mutation in sporadic BCP-ALL because 10 of 11 patients reported to have an R140L mutation in the literature also had the R38H mutation, which suggests a cooperation between these 2 mutations15 rather than just a biallelic inactivation of PAX5.16
The onset of BCP-ALL in patients with PAX5 G183S germline mutations located in the octapeptide domain is very early with a median age of 2 years, and it is associated with a loss of chromosome 9p, leading to the simultaneous losses of the second PAX5 allele and CDKN2A.6,7 In contrast, the germline R38H variant (in this study and in Yazdanparast et al8 ) is associated with an older age of onset (11 to 25 years old) and a normal karyotype (supplemental Figure 5; supplemental Table 5).
To functionally address the impact of the R38H mutation on B-cell differentiation, we transduced PAX5 wild-type (WT) and/or R38H murine fetal liver Pax5−/− B cells (Figure 2A) and the murine plasmacytoma 558LµM cell line that does not express Pax5 and Cd79a (supplemental Figure 6). As expected, PAX5 WT restored the expression of Cd19 in Pax5−/− cells and their capacity to differentiate in contrast to R38H (Figure 2B). R38H was also unable to rescue surface immunoglobulin M expression in the 558LµM cell line (supplemental Figure 6). In addition, R38H exhibited a weak competitive or additive effect on PAX5 WT when co-expressed in primary cells or in the 558LµM cell line, respectively (Figure 2B; supplemental Figure 6). PAX5 WT significantly increased the in vitro clonal activity of transduced Pax5−/− B220+ cells and led to the expression of Cd19 unlike R38H (supplemental Figure 7), confirming that R38H is not able to trigger B-cell differentiation. Furthermore, although no difference in frequencies of positive wells between PAX5 R38H- and MIG-infected cells was observed, the absolute number of B cells obtained per well was significantly higher (supplemental Figure 7). Altogether, our data demonstrate that R38H disables B-cell differentiation and partially maintains PAX5-dependent cell growth properties without overt dominant-negative effect on the normal PAX5 function.
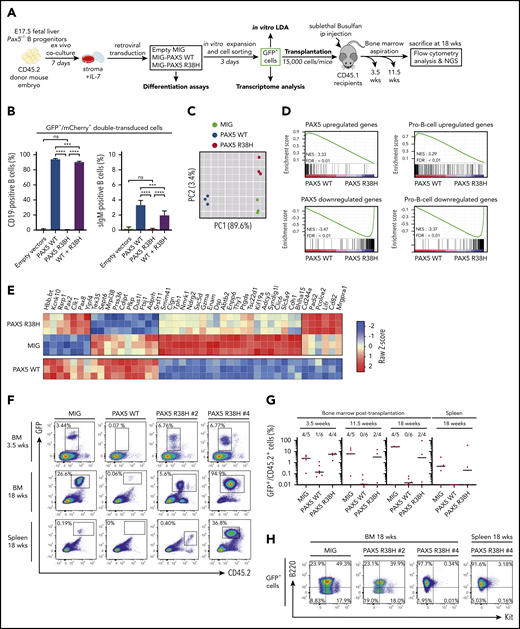
PAX5 R38H behaves as a hypomorphic variant and can predispose to BCP-ALL in mice. (A) Experimental scheme of retroviral complementation assays. (B) Pax5−/− cells were co-transduced with either empty MIG (MSCV-IRES-GFP) and MImCherry (labeled as empty vectors), empty MImCherry and MIG PAX5 WT (labeled as PAX5 WT), empty MImCherry and MIG-PAX5 R38H (labeled as PAX5 R38H), or MImCherry-PAX5 WT and MIG-PAX5 R38H (labeled as WT + R38H) retroviral vectors. The proportions of cell markers were evaluated by fluorescence-activated cell sorting (FACS) for each condition (n = 8 per condition). Data are representative of 2 independent experiments (n = 4 independent infections per experiment). Percentages indicate proportions of GFP/mCherry-double-positive that are CD19+ or surface IgM (sIgM)-positive as indicated. (C) Principal component analysis (PCA) of the 2 best components for the 2000 most differentially expressed genes among the 3 conditions. Green dots represent empty MIG condition, blue dots MIG-PAX5 WT condition, and red dots MIG-PAX5 R38H condition. (D) Gene set enrichment analysis of PAX5 target gene and pro-B cell gene sets17 in expression profiles of Pax5−/− cells transduced with PAX5 WT vs PAX5 R38H with false discovery rate (FDR) <0.01. (E) Comparative supervised heatmap using a Z-score and Spearman correlation clustering displaying the 44 most differentially expressed coding genes between PAX5 R38H-expressing Pax5−/− pro-B cells and MIG condition (fold >1.5; q < 0.05), with side comparison (lower panel) of corresponding gene expression in PAX5 WT-expressing Pax5−/− pro-B cells. (F) Representative FACS plots showing engraftment of the GFP+ donor cells in bone marrow (BM) samples at 3.5 and 18 weeks and spleen samples at 18 weeks posttransplantation. Mice were numbered with #x. (G) Quantification of engraftment of CD45.2+/GFP+ donor cells over time in BM at 3.5, 11.5, and 18 weeks posttransplantation and in spleen 18 weeks posttransplantation. Each dot represents individual mice (n = 4 to 6). Data show medians of engrafted mice (CD45.2+/GFP+ cell proportion >1% of total cells). (H) Representative FACS plots showing B220 (CD45R) and Kit (CD117) expression in CD45.2+/GFP+ cells of BM or spleen of PAX5 R38H- or MIG- transduced cells 18 weeks after transplantation. Mouse #4 shows BCP-ALL phenotype. ip, intraperitoneal; NES, normalized enrichment score; ns, not significant. Results are expressed as mean ± standard deviation. ***P < .001; ****P < .0001.
PAX5 R38H behaves as a hypomorphic variant and can predispose to BCP-ALL in mice. (A) Experimental scheme of retroviral complementation assays. (B) Pax5−/− cells were co-transduced with either empty MIG (MSCV-IRES-GFP) and MImCherry (labeled as empty vectors), empty MImCherry and MIG PAX5 WT (labeled as PAX5 WT), empty MImCherry and MIG-PAX5 R38H (labeled as PAX5 R38H), or MImCherry-PAX5 WT and MIG-PAX5 R38H (labeled as WT + R38H) retroviral vectors. The proportions of cell markers were evaluated by fluorescence-activated cell sorting (FACS) for each condition (n = 8 per condition). Data are representative of 2 independent experiments (n = 4 independent infections per experiment). Percentages indicate proportions of GFP/mCherry-double-positive that are CD19+ or surface IgM (sIgM)-positive as indicated. (C) Principal component analysis (PCA) of the 2 best components for the 2000 most differentially expressed genes among the 3 conditions. Green dots represent empty MIG condition, blue dots MIG-PAX5 WT condition, and red dots MIG-PAX5 R38H condition. (D) Gene set enrichment analysis of PAX5 target gene and pro-B cell gene sets17 in expression profiles of Pax5−/− cells transduced with PAX5 WT vs PAX5 R38H with false discovery rate (FDR) <0.01. (E) Comparative supervised heatmap using a Z-score and Spearman correlation clustering displaying the 44 most differentially expressed coding genes between PAX5 R38H-expressing Pax5−/− pro-B cells and MIG condition (fold >1.5; q < 0.05), with side comparison (lower panel) of corresponding gene expression in PAX5 WT-expressing Pax5−/− pro-B cells. (F) Representative FACS plots showing engraftment of the GFP+ donor cells in bone marrow (BM) samples at 3.5 and 18 weeks and spleen samples at 18 weeks posttransplantation. Mice were numbered with #x. (G) Quantification of engraftment of CD45.2+/GFP+ donor cells over time in BM at 3.5, 11.5, and 18 weeks posttransplantation and in spleen 18 weeks posttransplantation. Each dot represents individual mice (n = 4 to 6). Data show medians of engrafted mice (CD45.2+/GFP+ cell proportion >1% of total cells). (H) Representative FACS plots showing B220 (CD45R) and Kit (CD117) expression in CD45.2+/GFP+ cells of BM or spleen of PAX5 R38H- or MIG- transduced cells 18 weeks after transplantation. Mouse #4 shows BCP-ALL phenotype. ip, intraperitoneal; NES, normalized enrichment score; ns, not significant. Results are expressed as mean ± standard deviation. ***P < .001; ****P < .0001.
The transcriptome of R38H-transduced Pax5−/− cells (Figure 2C-E) is similar, although not identical, to the negative control (MIG) and clearly distinct from the PAX5 WT condition (Figure 2C). Gene set enrichment analyses confirmed that R38H is unable to regulate PAX5 target genes and to lead to a pro-B-cell gene expression program in contrast to PAX5 WT17 (Figure 2D; supplemental Figure 8). It is worth noting that the comparison of the residual transcriptional activity of PAX5 germline mutants showed that R38H has a stronger hypomorphic effect than G183S (supplemental Figure 9). Although the R38H transcriptome shares similarities with the one of PAX5 WT, supervised comparative analyses identified a molecular profile specific to R38H (Figure 2E). In particular, the expression of the leukemia inhibiting factor receptor (Lifr) gene18 implicated in stemness is downregulated in the PAX5 WT condition and upregulated in the R38H condition (Figure 2E). Our results suggest that PAX5 R38H confers some stemness features to the B cells. We thus transplanted transduced CD45.2+Pax5−/− B cells (Figure 2A,F-H). As previously described,19,20 MIG-transduced cells were able to home back to the bone marrow (BM) and to efficiently engraft at long term with an average of 30% CD45.2+/GFP+ cells in BM 18 weeks after transplantation (Figure 2F-G). In contrast, PAX5 WT–transduced cells abrogated their long-term engraftment capacity in the BM and spleen of recipient mice (Figure 2G). Interestingly, the ectopic expression of R38H led to an intermediate phenotype 3.5 weeks after transplantation and, as expected, none of the engrafted GFP+ cells acquired Cd19 (data not shown) because they do not express PAX5 WT. Interestingly, 2 of 4 mice maintained their engraftment 18 weeks posttransplantation and 1 (PAX5 R38H #4) developed a clonal B220+/IL7R+/Kit– leukemia (Figure 2H; supplemental Figure 10) with acquisition of a Jak3V670G mutation (supplemental Table 6).
In conclusion, we report a germline PAX5 p.R38H mutation in a family in which 3 children developed BCP-ALL. We demonstrated that R38H acts as a hypomorphic variant, does not abrogate the engraftment capacity of transduced Pax5−/− pro-B cells, and can predispose to BCP-ALL. The existence of asymptomatic patients without evidence of immunodeficiency before the onset of BCP-ALL shows that PAX5 germline cases might be underestimated and should be considered before any familial allograft.
Contact Cyril Broccardo (cyril.broccardo@inserm.fr) for original data.
The online version of this article contains a data supplement.
Acknowledgments
The authors thank all the patients and their families for consenting to participate in this study; Christophe Roumier (Tumor Bank, certification NF 96900-2014/65453-1, Centre Hospitalier Universitaire de Lille) for handling, conditioning, and storing patient samples; Meinrad Busslinger, MD, for the Pax5+/− mice and Michael Reth, for the 558LµM cells; Manon Farcé from the cytometry and cell sorting facility of the Technology Cluster of the Cancer Research Center of Toulouse (INSERM U1037) for technical assistance; and the Anexplo/Genotoul platforms for technical assistance (UMS006).
This work was supported by grants from the Ligue Nationale Contre le Cancer (R19015BB), the Association “les 111 des Arts” (R18062BB), the association "Capucine" and the Société Française des Cancers de l’Enfant (SFCE, R17094BB), the Région Occitanie (R16038BB), the Association “Cassandra” (R18041BB), and the Association “Constance La Petite Guerrière Astronaute” (R19043BB). L.A.J. was supported by grants from association "Capucine" and the SFCE (R17094BB) and Région Occitanie (RPH17006BBA).
Authorship
Contribution: N.D., L.A.J., B.G., E.D., C.P., and C. Broccardo conceived and designed the experiments; N.D., L.F., N.H., S.G., A.C., A.M.-R., C.R.-L., C.V., and M.F. performed molecular and cytogenetic analyses; L.A.J., C.H., S.H., and B.G. performed and analyzed in vivo and in vitro studies; N.P. and S.D. performed the microarray experiments; L.L. analyzed the mouse next-generation sequencing data; V.F. analyzed the transcriptomic data; S.L. provided genetic counseling; S.P., C. Berthon, B.N., and J.F. provided samples and clinical data; and N.D., L.A.J., B.G., E.D., C.P., and C. Broccardo wrote the manuscript with feedback from all authors.
Conflict-of-interest disclosure: The authors declare no competing financial interests.
Correspondence: Nicolas Duployez, Laboratory of Hematology, Biology and Pathology Center, Lille University Hospital, Boulevard Professeur Leclercq, 59037 Lille, France; e-mail: nicolas.duployez@chru-lille.fr; and Cyril Broccardo, Centre de Recherche en Cancérologie de Toulouse, INSERM U1037-CRCT, 2 Avenue Hubert Curien, 31037 Toulouse Cedex 01, France; e-mail: cyril.broccardo@inserm.fr.

This feature is available to Subscribers Only
Sign In or Create an Account Close Modal