

In this issue of Blood, report 6 unrelated male patients who carried gain-of-function (GOF) variants of the X-linked gene TLR8.1 The patients had invasive bacterial and fungal infections associated with splenomegaly and lymphadenopathy. They had an excess of double-negative T cells, abnormal B-cell maturation, and neutropenia, and some patients had bone marrow failure.
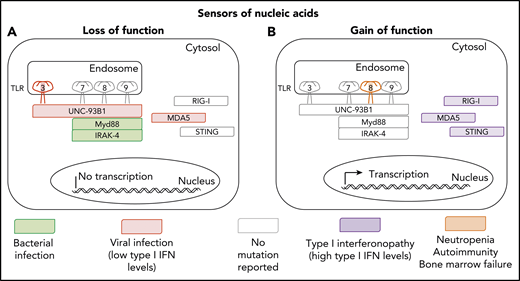
Inborn and somatic errors of immunity driven by nucleic acid sensors. (A) Clinical phenotypes associated with loss-of-function variants underlying autosomal dominant (TLR3) or autosomal recessive deficiencies (TLR3, UNC93B1, MYD88, IRAK-4, and MDA5). (B) Clinical phenotypes associated with gain-of-function variants underlying autosomal dominant disorders (RIG-I, MDA-5, STING, and TLR8).
Inborn and somatic errors of immunity driven by nucleic acid sensors. (A) Clinical phenotypes associated with loss-of-function variants underlying autosomal dominant (TLR3) or autosomal recessive deficiencies (TLR3, UNC93B1, MYD88, IRAK-4, and MDA5). (B) Clinical phenotypes associated with gain-of-function variants underlying autosomal dominant disorders (RIG-I, MDA-5, STING, and TLR8).
Human TLR8 is an endosomal type 1 transmembrane protein.2 On binding to single-strand RNA (ssRNA) viral intermediates or byproducts, it induces the production of proinflammatory cytokines and antiviral type 1 interferons (IFNs). Intriguingly, the mouse orthologue of TLR8 does not recognize human TLR8 ssRNA ligands, and its function remains elusive.3 Signal transduction via human TLR8 is dependent on UNC93B1, MyD88, and IRAK-4. TLR8 is expressed mostly on myeloid cells, including neutrophils, monocytes, macrophages, and conventional dendritic cells (mostly cDC2). Surprisingly, the GOF TLR8 variants do not seem to underlie a type 1 interferonopathy, contrasting with GOF mutations of genes encoding 3 other viral sensors, MDA5, RIG-I, and STING, which cause different forms of type 1 interferonopathy (see figure).4,5 Instead, GOF TLR8 variants underlie a series of surprising immunological and clinical phenotypes that raise multiple questions. What is the mechanism of neutropenia? It could be cell intrinsic, because TLR8 is expressed by neutrophils, but it could also be cell extrinsic, given the presence of autoantibodies against neutrophils. What about bone marrow failure? What are the mechanisms underlying T-cell proliferation and B-cell deficiency? These mechanisms are probably cell extrinsic, as TLR8 is not expressed by T and B cells, and they probably involve the production of cytokines other than type 1 IFNs by myeloid cells. What triggers the activation of the TLR8 GOF, which does not seem to be constitutive? Does the disease result from the gradual and cumulative effects of previous viral infections, or from persistent stimulation by endogenous agonists of TLR8?
Attesting to the pathogenic strength of the GOF variants, they were somatic, as opposed to germline, in 5 of the 6 patients. Somatic phenocopies of inborn errors of immunity are increasingly being recognized.6,7 The affected loci include CYBB, KRAS, NLRC4, NLRP3, NOD2, NRAS, STAT3, STAT5B, TMEM173, TNFRSF1A and TNFRSF6. Interestingly, GOF is the mechanism of “dominance” for 9 of these 11 genes (all except CYBB and STAT3). Moreover, in diseases related to somatic GOF, pathogenicity requires only 1% to 50% of leukocytes to carry the variant. The percentage of leukocytes carrying somatic GOF TLR8 variants ranges from 8% to 28%. Even a minor contingent of leukocytes hemizygous for a TLR8 GOF variant is sufficient to drive disease. The expression of GOF TLR8 by fewer than a third and perhaps even <10% of myeloid cells may therefore be sufficient for pathogenicity, which suggests that the mechanism of neutropenia involves a cell-extrinsic component, such as autoantibodies against neutrophils. It is important that the nature of the TLR8 alleles in all leukocyte subsets of the 5 patients with somatic variants be studied. At any rate, the small proportion of mutant leukocytes makes genetic detection for diagnosis more challenging. Deep sequencing of leukocytes, their subsets, or even single-cell sequencing, could provide a solution to this problem. The use of appropriate sources of DNA and computational pipelines is important, as these low-frequency reads are best searched for in leukocytes, and should not be filtered out, as is commonly the case for germline variants.
No disease has yet been attributed to TLR8 deficiency. These findings for TLR8 GOF variants suggest that patients with inherited TLR8 deficiency, if indeed there are any, may not display viral phenotypes, at odds with findings for patients with the various forms of inherited TLR3 deficiency, who have diverse viral diseases (see figure).8 TLR3 can be engaged by virus-triggered double-strand RNAs and elusive endogenous agonists.8 Previous descriptions of patients with inborn errors of components of the TLR7-TLR8-TLR9 pathway did not reveal an essential role of this pathway in host defense. Indeed, leukocytes from known patients with inborn errors of UNC93B do not respond to TLR3, TLR7, TLR8, and TLR9, and their viral phenotypes are caused by the disruption of the TLR3 pathway.9 Moreover, reported patients with MyD88 or IRAK4 deficiency, whose leukocytes respond to TLR3 but not to TLR7, TLR8, and TLR9 agonists, did not display severe viral illnesses.9 They had pyogenic bacterial infections caused by blunted interleukin-1R–mediated responses and responses to other TLRs.9 In light of the study by Aluri et al, the function of TLR8 in host defense remains mysterious, but we should perhaps be looking at patients with clinical phenotypes other than viral infections to resolve this enigma. The discovery of individuals with inherited TLR8 deficiency is awaited to clarify the function of this molecule, and it would not be surprising to encounter another surprise, so to speak.
Or could it be that the germline loss of TLR8 is neutral in humans? This is unlikely, as TLR8 shows very little allelic variability in human populations. Together with TLR3, TLR7, and TLR9, it is one of the TLRs under the strongest negative selection pressure,10 which indicates that any TLR8 genotype that damages the evolutionarily selected function of TLR8 confers a loss of fitness. It could be argued that this negative selection attests to the loss of fitness conferred by GOF variants, but this is highly unlikely, because it would require many, if not most of the missense variants to be GOF. However, 4 of the 6 patients reported by Aluri et al carried the same P432L GOF variant, strongly suggesting that most TLR8 missense variants are not GOF. Moreover, as out-of-frame TLR8 variants are rare in the general population, even in the heterozygous state in women, a more likely alternative is that negative selection at the TLR8 locus attests to a nonredundant but currently unknown role in host defense. As the infection profiles of known patients with UNC93B1 and MyD88/IRAK4 deficiencies who have been observed over the past 20 years do not seem to overlap, it is tempting to speculate that viral diseases of the past, such as the 1918 influenza (and, sadly, perhaps of today, such as COVID-19) may require TLR8 for its containment. However, it may well be that inherited TLR8 deficiency underlies another, nonviral clinical phenotype that may yet surprise us.
Conflict-of-interest disclosure: The authors declare no competing financial interests.

This feature is available to Subscribers Only
Sign In or Create an Account Close Modal