


Abstract
The core binding factor composed of CBFβ and RUNX subunits plays a critical role in most hematopoietic lineages and is deregulated in acute myeloid leukemia (AML). The fusion oncogene CBFβ-SMMHC expressed in AML with the chromosome inversion inv(16)(p13q22) acts as a driver oncogene in hematopoietic stem cells and induces AML. This review focuses on novel insights regarding the molecular mechanisms involved in CBFβ-SMMHC–driven leukemogenesis and recent advances in therapeutic approaches to target CBFβ-SMMHC in inv(16) AML.
Core binding factor in hematopoiesis
The core binding factor (CBF) is a heterodimeric transcription factor complex composed of 2 core subunits: DNA-binding RUNX protein encoded by 1 of 3 genes (RUNX1, RUNX2, and RUNX3) and the non–DNA-binding partner CBFβ.1,2 The function of CBFβ in hematopoiesis is to enhance DNA binding and protein stability of RUNX proteins. Runx1−/− and Cbfb−/− embryos display a profound block in definitive hematopoiesis and die at E11.5-12.5.3-7 Runx1 is required for the transition from hemogenic endothelial cell to hematopoietic stem cell (HSC) during development, but it is dispensable for continued fetal hematopoiesis after HSC formation occurs.5 Similar to Runx1, Cbfβ is essential for the transition from endothelial cell to HSC during embryonic development. Although restoration of Cbfβ within the endothelium and HSCs of Cbfb−/− embryos rescued fetal liver hematopoiesis, these embryos displayed lack of hematopoietic maturation in both myeloid and lymphoid lineages and defective bone formation, resulting in death at birth.8 Thus, although Runx1 and Cbfβ are both essential for HSC formation in the early stage of definitive hematopoiesis, only Cbfβ is required for differentiation of myeloid and lymphoid lineage cells in the consecutive stages of embryonic hematopoiesis. Because Cbfβ has also been shown to form heterodimers with Runx2 and Runx3, which are expressed in hematopoietic cells,9-11 it is possible that Runx2 and Runx3 functions are impaired in the absence of Cbfβ. Adult Runx1 and Cbfb conditional knockout mice displayed expansion of the hematopoietic stem and progenitor cell pool and defects in differentiation into megakaryocytes, T- and B-lymphocytes, and platelets.12-15 Collectively, both RUNX1 and CBFβ play major roles in an overlapping and stage-specific manner in embryonic and adult hematopoiesis. Whether interaction with other transcription factors and chromatin regulators16,17 is contributing to distinct functions for RUNX1 and CBFβ during multiple developmental stages awaits further experiments.
inv(16) AML
The inv(16)(p13q22) and the less frequent t(16;16)(p13q22) result in the formation of a chimeric gene consisting of the 5′ portions of CBFB fused to the 3′ portion of the smooth muscle myosin heavy chain gene MYH11 (Figure 1).18,19 The resulting CBFB-MYH11 fusion gene encodes the fusion protein CBFβ-SMMHC. The inv(16) mutation is reported in 5% to 8% of patients with AML.20 AML blasts with inv(16) have a myelomonocytic morphology with immature basophilic granules and are classified as AML subtype M4-with eosinophilia (M4-eo).21
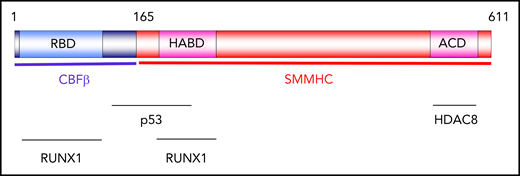
Distribution of CBFβ-SMMHC functions. Schematic representation of functional domains of CBFβ-SMMHC. The RUNX1-binding domain (RBD) and high-affinity–binding domain (HABD) mediate interaction with RUNX1. The assembly-competence domain (ACD) in SMMHC mediates dimerization and multimerization. ACD is also required for interaction with histone deacetylase HDAC8. The region located between amino acids 134 and 221 mediates interaction with p53.
Distribution of CBFβ-SMMHC functions. Schematic representation of functional domains of CBFβ-SMMHC. The RUNX1-binding domain (RBD) and high-affinity–binding domain (HABD) mediate interaction with RUNX1. The assembly-competence domain (ACD) in SMMHC mediates dimerization and multimerization. ACD is also required for interaction with histone deacetylase HDAC8. The region located between amino acids 134 and 221 mediates interaction with p53.
RUNX1 binds to CBFβ-SMMHC through 2 domains: the RUNX-binding domain (RBD) in the CBFβ part, and the high-affinity–binding domain (HABD) in the N-terminal portion of SMMHC (Figure 1).22 RUNX1 has a 10-fold higher affinity to CBFβ-SMMHC than it has to CBFβ.22 The assembly-competence domain (ACD) at the C-terminus of SMMHC is critical for dimerization and multimerization of CBFβ-SMMHC.23 CBFβ-SMMHC multimerization is essential for the localization of CBFβ-SMMHC to the nucleus and for its leukemic activity.24,25 CBFβ-SMMHC interacts with p53 through a site spanning the junction between CBFβ and SMMHC.26 ACD interacts with histone deacetylase 8 (HDAC8),27 and this interaction is critical for CBFβ-SMMHC–induced p53 deacetylation and leukemia induction.26 Whether RUNX2 and RUNX3 bind to CBFβ-SMMHC remains unknown.
Mechanism of CBFβ-SMMHC function in AML
CbfbMYH11/+ knock-in embryos demonstrated a loss of definitive hematopoiesis and embryonic lethality at E12.5.28 In adult hematopoiesis, CBFβ-SMMHC expression deregulates multilineage hematopoietic differentiation and induces AML.29,30 Because CbfbMYH11/+ mouse model phenotypes are consistent with the knockout mouse model for Runx1, it was suggested that CBFβ-SMMHC exerts dominant negative function over RUNX1. One characteristic of inv(16) leukemia is the presence of abnormal myeloid progenitors (AMPs), which are known to have preleukemic potential and to display a hybrid myeloid-erythroid progenitor and common myeloid progenitor immunophenotype.30,31 Even though inv(16) is known to act as a founder mutation, secondary mutations in genes that activate tyrosine kinase signaling (such as KIT, NRAS, KRAS, and FLT3) are required for the initiation of leukemia.31-35
Whether RUNX1 protein per se is required for inv(16) leukemogenesis remains contentious. Mice that express the conditional CbfbMYH11/+ knock-in allele in a conditional Runx1 knockout background failed to develop AML,36 which suggests that Runx1 is required for the development of leukemia. RUNX1 expression is required for inv(16) AML cells to survive in culture.37 There are many explanations for why CBFβ-SMMHC is dependent on RUNX1. CBFβ-SMMHC recruitment to DNA is mostly RUNX1 dependent, and CBFβ-SMMHC functions in association with RUNX1 to control gene expression.36,38-40 RUNX1 is required for the development of preleukemic AMPs in inv(16) AML.36 So, it is possible that in the absence of RUNX1, CBFβ-SMMHC is unable to associate with DNA and fails to regulate the expression of leukemic-specific genes, which in turn hinders AMP formation and thereby prevents the development of AML. Second, initiation of myeloid leukemia occurs within a window of differentiation in the myeloid lineage, and granulocyte-macrophage progenitors provide a genomic environment that permits AML transformation.41-43 RUNX1 directly regulates C/EBPα,44 a master regulator of granulocyte differentiation,45 and Runx1 haplo-insufficient mice display defects in granulocyte maturation.46 Therefore, disruption of granulocyte differentiation due to the loss of Runx1 could be playing a role in the delay in leukemia development in the context of CBFβ-SMMHC.
Loss of RUNX1 function by either RUNX1 point mutations or translocation t(8;21) in AML present with an undifferentiated or myeloblastic differentiation phenotype (French-American-British Classification M0 and M2, respectively). However, inv(16) AML has a further differentiated myelomonocytic state (M4Eo). Gene expression profiling of inv(16) AML cells displays a pattern distinct from that of t(8;21) AML.47 Comparison of transcriptional changes in lineage-negative Sca-1+c-Kit+ bone marrow cells in adult Runx1 conditional knockout mice14 with lineage-negative c-Kit+ bone marrow cells in adult CbfbMYH11/+ conditional knock-in mice48 demonstrated that only 24% of differentially regulated genes in Runx1 knockout mice are also deregulated in CbfbMYH11/+ knock-in mice (J.A.P., unpublished observation). In addition to RUNX1, CBFβ-SMMHC interacts with several key hematopoietic transcription factors and chromatin regulators and co-occupies several promoters.39,40 Collectively, these findings suggest that CBFβ-SMMHC has a major impact on leukemogenesis independent of RUNX1 inhibition.
Recent advances in inhibiting CBFβ-SMMHC activity
Adults with inv(16) AML have traditionally received 7+3 style induction chemotherapy with infusional cytarabine and anthracycline. These regimens are effective at achieving complete remission in more than 90% of patients with inv(16) AML.49 However, the 3-year disease-free survival of adults older than age 35 years is 30% as opposed to 67% for those younger than age 35 years.49 Older adults with inv(16) AML are less able to tolerate higher-intensity postremission consolidation chemotherapy regimens, and they have a higher incidence of additional genetic lesions accompanying the inv(16).49,50 The last decade saw several clinical advances, including the approval of 8 novel agents by the US Food and Drug Administration for treating AML.51-53 The most noteworthy for inv(16) AML is the addition of gemtuzumab ozogamicin (GO), an anti-CD33 monoclonal antibody, to induction chemotherapy. The ALFA-0701 trial demonstrated that the addition of GO to induction chemotherapy significantly improved outcome in patients with inv(16) AML.54-56 The fludarabine, cytarabine, and granulocyte colony-stimulating factor with GO (FLAG-GO) regimen may provide additional overall survival and relapse-free survival benefit for patients with inv(16) AML.57 KIT mutations are associated with early relapse and high risk in inv(16) AML.58,59 Incorporation of dasatinib (the tyrosine kinase inhibitor with KIT activity) improved disease-free survival for patients with inv(16) AML.60,61 Venetoclax, the BCL2 inhibitor, in combination with a hypomethylating agent has shown encouraging response rates for patients with AML, including those with inv(16).62,63 More detailed studies are required to determine the usefulness of dasatinib and venetoclax for treating patients with inv(16) AML. Although survival of patients with inv(16) AML is higher than that for patients with other AML subtypes, 30% to 40% of patients still relapse and die of the disease,64,65 suggesting the need to develop better therapeutic strategies for treating patients with inv(16) AML.
The inv(16) mutation arises as a founder mutation in HSCs and produces preleukemic HSCs associated with a block in myeloid differentiation that eventually induces AML.66 Inhibiting secondary mutations and their associated pathways is less likely to provide greater clinical outcome because of the lack of targeting preleukemic clones that resist standard chemotherapy, which in turn predispose to relapse. Meanwhile, targeting CBFβ-SMMHC warrants greater therapeutic benefit since it is the founder mutation present in all preleukemic clones. Here we discuss recent developments in inhibiting CBFβ-SMMHC function for inv(16) AML treatment.
By using a fluorescence resonance energy transfer (FRET) assay for compounds that inhibit RUNX1 and CBFβ protein-protein interaction, Cunningham et al67 identified Ro5-3335 as a small molecule inhibitor of RUNX1-CBFβ protein-protein interaction (Figure 2A). Ro5-3335 reduced the viability of RUNX1-ETO (the t(8;21) fusion oncoprotein) and CBFβ-SMMHC AML cells and reduced leukemia burden in CbfbMYH11/+ knock-in mice. Ro5-3335 binds to bromodomain protein WDR9 and the SWI-SNF chromatin remodeling complex subunit SMARCA2 and inhibits their function.68 Because WDR9 (also known as BRWD1) and SWI-SNF complex are known to play roles in chromatin regulation in cooperation with RUNX1,69,70 it is highly possible that the anti-leukemic effects of Ro5-3335 are due to indirect RUNX1 inhibition.
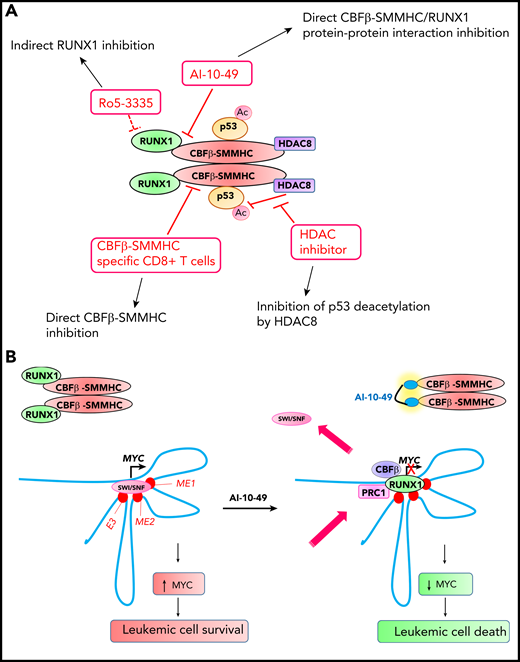
Novel therapeutic approaches for inhibiting CBFβ-SMMHC in inv(16) AML. (A) Small molecule and T-cell immunotherapy strategies for inhibiting CBFβ-SMMHC and their mechanisms of action. Ac, acetylation. (B) RUNX1-MYC axis in inv(16) AML cell survival. CBFβ-SMMHC tethers RUNX1, and this results in MYC activation and enhanced survival in inv(16) AML cells. Treatment of inv(16) AML cells with AI-10-49 induces an acute release of RUNX1 from CBFβ-SMMHC, increases RUNX1 occupancy at the MYC distal enhancers, replaces the SWI-SNF chromatin remodeling complex, and recruits polycomb-repressive complex 1 (PRC1), which in turn induces apoptosis by repressing MYC. E3, +1.7 Mb BRD4-mediated MYC enhancer (BDME)80 element 3; ME1, +0.18 Mb MYC enhancer 1; ME2, +0.5 Mb MYC enhancer 2.
Novel therapeutic approaches for inhibiting CBFβ-SMMHC in inv(16) AML. (A) Small molecule and T-cell immunotherapy strategies for inhibiting CBFβ-SMMHC and their mechanisms of action. Ac, acetylation. (B) RUNX1-MYC axis in inv(16) AML cell survival. CBFβ-SMMHC tethers RUNX1, and this results in MYC activation and enhanced survival in inv(16) AML cells. Treatment of inv(16) AML cells with AI-10-49 induces an acute release of RUNX1 from CBFβ-SMMHC, increases RUNX1 occupancy at the MYC distal enhancers, replaces the SWI-SNF chromatin remodeling complex, and recruits polycomb-repressive complex 1 (PRC1), which in turn induces apoptosis by repressing MYC. E3, +1.7 Mb BRD4-mediated MYC enhancer (BDME)80 element 3; ME1, +0.18 Mb MYC enhancer 1; ME2, +0.5 Mb MYC enhancer 2.
Qi et al26 evaluated the therapeutic benefit of inhibiting HDAC8 in inv(16) AML. CBFβ-SMMHC recruits HDAC8 and p53 in a protein complex and promotes deacetylation of p53 by HDAC8 (Figure 2A). HDAC8 inhibition by small molecule inhibitors prevents p53 inactivation by HDAC8, thereby inducing p53-mediated apoptosis in inv(16) leukemic cells and extending survival in the CbfbMYH11/+ knock-in mouse model, suggesting the potential of using HDAC8 inhibition for treating inv(16) AML.
Attempts to directly inhibit CBFβ-SMMHC-RUNX1 were reported by Illendula et al.71 By using a FRET assay for compounds that inhibit CBFβ-SMMHC and RUNX1 protein-protein interaction, AI-4-57 was discovered as the lead compound, which was modified to AI-10-47 for better pharmacokinetics. To specifically inhibit CBFβ-SMMHC-RUNX1, principles of polyvalency, which state that the affinity of polyvalent ligands is greater than the sum of monovalent affinities,72 were applied. CBFβ exists in a monomeric state and CBFβ-SMMHC exists in an oligomeric state. So, according to the polyvalency principle, a dimeric version of AI-10-47 should bind with higher affinity to CBFβ-SMMHC, with little binding to CBFβ. Multiple dimeric compounds with varying linker length were synthesized, and AI-10-49, a molecule with high sensitivity to CBFβ-SMMHC-RUNX1, was developed. Thus, AI-4-57 was structurally modified at 3 levels for improved pharmacokinetics, specificity, and sensitivity, which made AI-10-49 highly potent. AI-10-49 specifically inhibits CBFβ-SMMHC-RUNX1 protein-protein interaction without inhibiting CBFβ-RUNX1 (Figure 2A). AI-10-49 significantly extended survival of mice transplanted with inv(16) leukemic cells without any toxic effects. AI-10-49 treatment in inv(16) AML cells results in a marked repression of the MYC signature, and MYC silencing induces apoptosis in inv(16) leukemic cells.73 AI-10-49 treatment increases RUNX1 occupancy at 3 MYC distal enhancers and disrupts chromatin dynamics, which in turn induces apoptosis by repressing MYC (Figure 2B). Pharmacologic inhibition of MYC activity using a combined treatment with AI-10-49 and the bromodomain inhibitor JQ1 revealed a strong synergy and a significant delay in leukemia latency in mice, providing proof-of-principle for the usefulness of combined therapy in inv(16) AML. AI-10-49 has been licensed to Systems Oncology for further drug development.
Little is known about the efficacy of T-cell immunotherapy for AML treatment. A recent study tested the immunogenicity of candidate CBFβ-MYH11 peptide epitopes in culture by stimulating HLA-typed healthy donor CD8+ T cells with autologous mature dendritic cells pulsed with a pool of candidate peptides (Figure 2A).74 High-avidity CD8+ T-cell clones specific for the HLA-restricted CBFβ-SMMHC epitope were able to lower disease burden in vivo in a patient-derived murine xenograft model, suggesting that CBFβ-SMMHC is a bona fide target for T-cell immunotherapy.
Future directions
Treatment for AML had not changed much in decades until recently.51 AI-10-49 serves as a proof-of-concept for targeted therapies for inv(16) AML.75 Although inhibition of ribosome biogenesis in inv(16) AML cells treated with AI-10-49 is due to MYC73 (a major player in ribosome biogenesis76 ), CBFβ-SMMHC was reported to have a direct role in regulating ribosome biogenesis.77 Further investigations regarding inhibition of ribosome biogenesis are likely to provide additional therapeutic opportunities. Future studies using combinations of AI-10-49 with HDAC8 inhibitors as well as chemotherapeutic agents should lead to greater therapeutic responses for inv(16) AML. An innovative concept that is gaining attention in the drug discovery field is the advent of proteolysis-targeting chimeras (PROTACs), small molecules that concurrently bind to a target protein and an E3 ubiquitin ligase, thereby resulting in ubiquitination and protein degradation.78,79 Further research aimed at developing CBFβ-SMMHC–specific PROTACs may have potential as therapy for inv(16) AML.
Acknowledgments
The authors thank Sridhar Rao, Karen Sue Carlson, and Nan Zhu for critically reviewing the manuscript.
This work was supported by a grant from the Versiti Blood Research Institute Foundation (J.A.P.), by grants from the National Institutes of Health (NIH) National Cancer Institute (R35CA197697) (D.G.T.) and the NIH National Heart, Lung, and Blood Institute (P01HL131477) (D.G.T.), and from the Singapore Ministry of Health (STaR18nov-0002).
Authorship
Contribution: S.S., D.G.T., and J.A.P. wrote the manuscript.
Conflict-of-interest disclosure: J.A.P. holds a patent for AI-10-49 (US2019/033889). The remaining authors declare no competing financial interests.
Correspondence: John A. Pulikkan, Program in Stem Cell Biology and Hematopoiesis, Versiti Blood Research Institute, 8733 W Watertown Plank Rd, Milwaukee, WI 53226; e-mail: jpulikkan@versiti.org; and Daniel G. Tenen, Cancer Science Institute, National University of Singapore, Singapore 117599, Singapore; e-mail: csidgt@nus.edu.sg.
