

Key Points
Natural CXCR4 mutants show subset-specific effects of CXCR4 signaling on the dynamics of DC migration in vivo.
Proper CXCR4 signaling directs efficient skin DC migration to lymph nodes, thus controlling their activation state.

Abstract
Dendritic cells (DCs) encompass several cell subsets that collaborate to initiate and regulate immune responses. Proper DC localization determines their function and requires the tightly controlled action of chemokine receptors. All DC subsets express CXCR4, but the genuine contribution of this receptor to their biology has been overlooked. We addressed this question using natural CXCR4 mutants resistant to CXCL12-induced desensitization and harboring a gain of function that cause the warts, hypogammaglobulinemia, infections, and myelokathexis (WHIM) syndrome (WS), a rare immunodeficiency associated with high susceptibility to the pathogenesis of human papillomavirus (HPV). We report a reduction in the number of circulating plasmacytoid DCs (pDCs) in WHIM patients, whereas that of conventional DCs is preserved. This pattern was reproduced in an original mouse model of WS, enabling us to show that the circulating pDC defect can be corrected upon CXCR4 blockade and that pDC differentiation and function are preserved, despite CXCR4 dysfunction. We further identified proper CXCR4 signaling as a critical checkpoint for Langerhans cell and DC migration from the skin to lymph nodes, with corollary alterations of their activation state and tissue inflammation in a model of HPV-induced dysplasia. Beyond providing new hypotheses to explain the susceptibility of WHIM patients to HPV pathogenesis, this study shows that proper CXCR4 signaling establishes a migration threshold that controls DC egress from CXCL12-containing environments and highlights the critical and subset-specific contribution of CXCR4 signal termination to DC biology.
Introduction
Dendritic cells (DCs) are a heterogeneous group of innate cells that control tolerance and immunity.1 They encompass plasmacytoid DC (pDC) and type 1 and 2 conventional DCs (cDCs), identified in humans and mice based on ontogenic, phenotypic, and functional criteria.2-5 DCs originate in the bone marrow (BM), either as fully differentiated pDCs, arising from both lymphoid and myeloid progenitors,5-7 or as myeloid-derived cDC precursors (pre-cDCs), which complete their differentiation in peripheral tissues or lymphoid organs (LOs).3,8 Langerhans cells (LCs)9 are an additional DC-like subset that collaborate with bona fide DCs to shape immune responses.
pDCs are circulating sentinels10 that promote antiviral immunity by type I interferon (IFN) secretion. In peripheral tissues, cDCs and LCs collect antigens before trafficking to secondary LOs (SLOs) as migratory DCs (migDCs) and migratory LCs, where they reach the T-cell zone to prime adaptive immune responses together with resident DCs (resDCs) and pDCs.11,12 The appropriate pDC and pre-cDC distribution throughout the tissues, as well as cDC and LC patrolling and directed migration to lymph nodes (LNs) warrant DC function.13,14 Although CCR7 and its ligands, CCL19 and CCL21, form the main chemokine (CK) receptor-CK axis that controls DC migration to and within SLOs,12,15-17 additional CKs and their receptors may regulate their location,10,12 including CXCR4 and its sole CK ligand, CXCL12.11,12,18-20
CXCR4 is a broadly expressed Gαi protein-coupled receptor,11 including in DCs, of which activation by CXCL12 is essential for myelopoiesis.21 CXCL1222 is highly produced by dermal fibroblasts, BM-derived mesenchymal cells, endothelial cells, and DCs.23-28 Although studies based on knock-out models and pharmacological blockade have revealed a role for CXCR4 in pDC differentiation,29,30 pDC and pre-cDC retention in the BM,31,32 and pDC,33 LC,34 and cDC migration,11,26 the true contribution of its signaling in DC subset homeostasis and function remains elusive. This can be addressed using natural CXCR4 mutants that cause the warts, hypogammaglobulinemia, infections, and myelokathexis (WHIM) syndrome (WS). This rare immunodeficiency mainly results from heterozygous CXCR4 mutations, leading to CXCR4 carboxyl-terminal domain truncation and subsequent gain-of-function and impaired desensitization (ie, dysfunction).25,35,36 WS manifests by panleukopenia37,38 and profuse intractable human papillomavirus (HPV)-induced warts that often evolve toward carcinoma.25,39-43 In this context, Tassone et al reported quantitative circulating DC defects in WHIM patients.44 However, the genuine impact of CXCR4 dysfunction on DC-subset differentiation, migration, and function, and thus the role of proper CXCL12/CXCR4 signaling in DC biology, have not been yet investigated.
Here, we used WS-associated CXCR4 mutations to address this issue. Our results in WHIM patients challenge those of a previous report,44 as we show quantitative defects for circulating pDC but not cDC subsets. They were confirmed in Cxcr4+/1013 knock-in mice that replicate WS-associated immune-hematological defects.25,45 In these mice, we show that pDC differentiation and function are preserved and that the pDC peripheral defect can be explained by their entrapment in the BM. Although LCs and dermal DCs were quantitatively preserved in Cxcr4+/1013 mice, their migration to LNs was markedly reduced, both at steady state and in a model of HPV-induced dysplasia. In addition, Cxcr4+/1013 migDCs (migDC+/1013) recovered from LNs were more activated than their wild-type (WT) counterparts, likely because of the selective egress of only the most activated skin DC+/1013. Overall, our results unravel the specific importance of proper CXCR4 signaling in DC-subset biology and its profound impact on immune homeostasis.
Methods
Human samples
WHIM patients were recruited from the French Severe Chronic Neutropenia Registry (J.D.), approved by the “Commission Nationale de l’Informatique et des Libertés.” The patients provided written informed consent for genetic testing and inclusion in the registry. The patients and their genetic mutations (supplemental Table 1, available on the Blood Web site) have been described.41,46
Blood samples were collected in lithium-heparin–coated tubes. For each patient sample, a sample from a healthy donor was collected at the same time and place for parallel analysis.
Mice
Cxcr4+/1013 mice harboring the WS-associated CXCR41013 mutation on the C57BL/6J and FVB/N genetic backgrounds were previously described.45,47 K14-HPV16 (HPV) transgenic mice expressing HPV early genes under the control of the keratin 14 promoter48,49 were obtained from the National Cancer Institute mouse repository. K14-HPV16 Cxcr4+/1013 (HPV+/1013) mice were generated by crossing FVB/N Cxcr4+/1013 mice with HPV mice.47
Except for parabiosis experiments, experimental procedures were conducted in the AnimEx animal facility (agreement no. B 92-023-01) in accordance with European Union legislation concerning the use of laboratory animals and approved by the Institutional Animal Care and Ethics Committee CEEA26 (Institut Gustave Roussy, France). Data were obtained from 7- to 14-week-old mice. Cxcr4+/1013 and HPV+/1013 mice were compared with their littermate Cxcr4+/+ and HPV controls.
Parabiosis experiments were performed in the Biological Resource Centre of A*STAR, under the authorization of the Institutional Animal Care and Use Committee of the Biological Resource Centre, in accordance with the guidelines of the Agri-Food and Veterinary Authority and the National Advisory Committee for Laboratory Animal Research of Singapore.
Parabiosis
Female CD45.2 Cxcr4+/1013 and CD45.1 control mice were anesthetized using 2.5% avertin (15 mL/kg) and surgically joined, as previously described.50 Mice then received Baytril and buprenorphine subcutaneously (0.05-2 mg/kg and 5-20 mg/kg, respectively). After 8 weeks, blood was recovered from each parabiont. Spleens and BM were harvested after perfusion. The blood volume present in each individual mouse was calculated as follows: blood volume (mL) = 0.0715 × body weight (g).51 Spleen/blood ratios were calculated as the number of cells in nonhost organ divided by the number of cells in nonhost blood, as previously described.50
In vivo mobilization
Mice were injected intraperitoneally with AMD3100 (Sigma-Aldrich) or phosphate-buffered saline. Two hours later, blood was collected for flow cytometry.
FITC painting assays
A solution of fluorescein isothiocyanate (FITC) (1 mg/mL in acetone; Merck) was mixed (v/v) with dibutyl phthalate and 100 µL applied to the abdomen. Mice were euthanized 24 hours later and migration of skin DCs to draining lymph nodes (SDLNs) was assessed by flow cytometry. For CXCR4 blockade, AMD3100 (0.8 mg/kg) was injected intraperitoneally at t = 0 and t = 6 hours. FITC application was performed 1 hour after the first AMD3100 injection.
Mouse samples
Epidermal sheets
Epidermal sheets were prepared as previously described.52 Details are provided in supplemental Methods.
DC activation
For in vivo stimulation, mice were injected intraperitoneally with 20 µg CpG-ODN 2216 (Invivogen) in a complex with 30 µg DOTAP liposome (Sigma-Aldrich), as previously described.56 Control mice received DOTAP only.
In vitro activation is detailed in supplemental Methods.
Cytokine quantification
The IFN-α concentrations in cell supernatants and CCL2, CCL4, and CXCL10 concentrations in mouse serum were quantified using a Mouse IFN α Platinum enzyme-linked immunosorbent assay (eBioscience) and U-PLEX Chemokine Combo Mouse Kit (Meso Scale Discovery), respectively.
Flow cytometry
Flow cytometry was performed as described in supplemental Methods.
Histology
Histology was performed as described in supplemental Methods.
Statistics
Statistical analyses were performed using Prism software (GraphPad). The 2-tailed Mann-Whitney test and paired t test were used to compare unpaired and paired data, respectively.
Results
WHIM patients display quantitative defects in circulating pDCs but not cDCs
We analyzed cDC1, cDC2, and pDC subsets in the blood of 4 previously described WHIM patients and 9 healthy controls. Patients were panleukopenic, as expected (Figure 1A).41 Blood pDCs counts and frequencies were reduced (Figure 1B-C), as previously reported.44 However, cDC1 and cDC2 frequencies were slightly higher and normal, respectively. These DC subsets showed no significant quantitative defects (Figure 1B-C), in contrast to the reduced percentages and counts reported by Tassone et al for the entire myeloid DC subset.44 Thus, WS-associated CXCR4 mutations were associated with a quantitative defect of circulating pDC but not cDC subsets in the blood of the 4 analyzed patients.
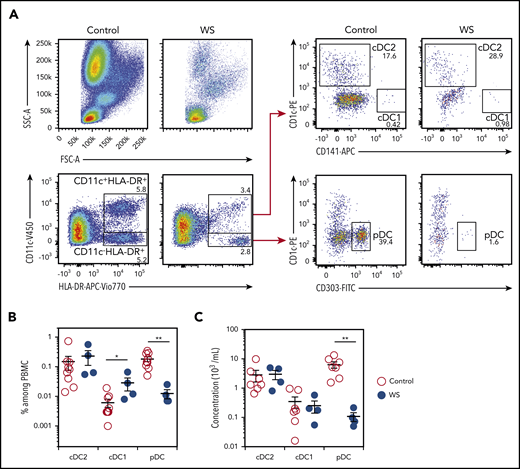
WHIM patients harbor quantitative defects in pDCs. (A) Blood DC subsets were analyzed in WHIM patients and healthy donors. Representative pseudo-color dot plots obtained for the same volume of blood from 1 healthy control (control) and 1 WHIM patient (WS) carrying the heterozygous CXCR4+/1013 mutation are shown. cDC2 was defined as CD11c+HLA-DR+CD1c+ cells, cDC1 as CD11c+HLA-DR+CD141+ cells, and pDCs as CD11c−HLA-DR+CD303+ cells. Percentages among parent cells are indicated. (B-C) The frequency of DC subsets among peripheral blood mononuclear cells (B) and their concentrations (C) were measured for healthy controls (control) and WHIM patients (WS). Each point represents an individual and lines indicate the mean ± standard error of the mean (SEM). n = 9 Controls and n = 4 WS (B); n = 7 controls and n = 4 WS (C). Statistical analysis was performed using the 2-tailed, unpaired Mann-Whitney test. *P < .05, **P < .01.
WHIM patients harbor quantitative defects in pDCs. (A) Blood DC subsets were analyzed in WHIM patients and healthy donors. Representative pseudo-color dot plots obtained for the same volume of blood from 1 healthy control (control) and 1 WHIM patient (WS) carrying the heterozygous CXCR4+/1013 mutation are shown. cDC2 was defined as CD11c+HLA-DR+CD1c+ cells, cDC1 as CD11c+HLA-DR+CD141+ cells, and pDCs as CD11c−HLA-DR+CD303+ cells. Percentages among parent cells are indicated. (B-C) The frequency of DC subsets among peripheral blood mononuclear cells (B) and their concentrations (C) were measured for healthy controls (control) and WHIM patients (WS). Each point represents an individual and lines indicate the mean ± standard error of the mean (SEM). n = 9 Controls and n = 4 WS (B); n = 7 controls and n = 4 WS (C). Statistical analysis was performed using the 2-tailed, unpaired Mann-Whitney test. *P < .05, **P < .01.
Cxcr4+/1013 mice reproduce WS-associated DC-subset alterations
We next analyzed DC subsets in Cxcr4+/1013 mice, which harbor the WS-associated CXCR41013 gain-of-function mutation25 and model the panleukopenia reported in patients.45 Cxcr4+/1013 mice showed reduced splenic pDC frequency and cellularity (Figure 2A-C) and reduced splenocyte numbers (supplemental Figure 1A).45 In contrast, the frequency of splenic Cxcr4+/1013 cDCs (cDC+/1013) was significantly higher, resulting in preservation of their number (Figure 2B-C), with no alteration in the relative proportion of cDC1s and cDC2s (supplemental Figure 1B). Cxcr4+/1013 pDC (pDC+/1013) expressed CD4, CD8α, CD9, and CCR9 similarly to control pDCs (pDCWT), but included a subset of cells expressing low levels of SiglecH (supplemental Figure 2A), which may correspond to myeloid pDC-like cells.7 pDC+/1013 and cDC+/1013 displayed enhanced chemotaxis toward CXCL12 in a transwell assay (supplemental Figure 2B), which was blocked by the CXCR4-specific antagonist AMD3100, with no alteration in CXCR4 cell-surface expression (supplemental Figure 2C). Further analyses in the LNs and thymus showed a lower number and frequency of pDC+/1013 than pDCWT, whereas LN resDC+/1013 and thymic cDC+/1013 frequencies were preserved (Figure 2D-I; supplemental Figure 1C-D). The blood pDC+/1013 frequency and counts were also reduced and pre-cDC+/1013 counts were diminished (supplemental Figure 3). Thus, Cxcr4+/1013 mice display alterations in DC-subset distribution consistent with those observed in WHIM patients, including a marked quantitative deficit of pDCs.
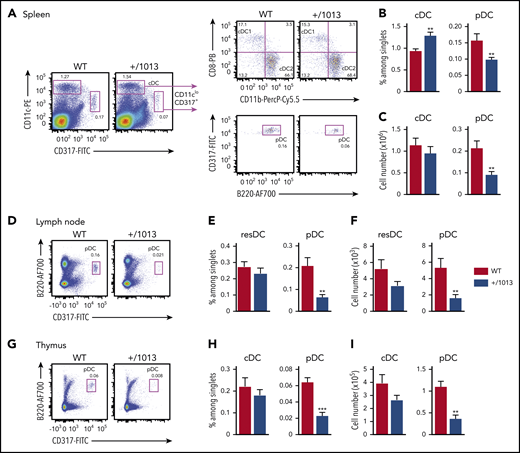
Cxcr4+/1013 mice show a reduced pDC pool in the spleen, LNs, and thymus. (A) Representative dot plots show the gating strategy for cDC1 (CD11chiCD317−CD11b−CD8+), cDC2 (CD11chiCD317−CD11b+CD8−), and pDC (CD11cloCD317+B220+) identification in the spleens from Cxcr4+/1013 (+/1013) and littermate Cxcr4+/+ (WT) mice. The percentages of cDCs, CD11cloCD317+ cells, and pDCs among singlets and cDC1 and cDC2 among DCs are indicated. (B-C) Spleen cDCs and pDCs frequencies (B) and numbers (C) were determined by flow cytometry (n = 17 mice/group from 7 experiments). (D,G) Representative dot plots showing pDC gating in inguinal LNs (D) and the thymus (G) from +/1013 and WT mice. (E-F) Resident cDC (resDC, CD11chiCD317−MHCII+) and pDC (CD11cloCD317+B220+) frequencies (E) and numbers (F) in inguinal LNs from +/1013 and WT mice (n = 12-13 mice/group from 5 experiments). (H-I) cDC (CD11chiCD317−) and pDC (CD11cloCD317+B220+) frequencies (H) and numbers (I) in thymuses from +/1013 and WT mice (n = 7-8 mice/group from 3 experiments). Bar graphs show the mean ± SEM. Mice had a C57BL/6J genetic background. Statistical analysis was performed using the 2-tailed, unpaired Mann-Whitney test. *P < .05, **P < .01, ***P < .001.
Cxcr4+/1013 mice show a reduced pDC pool in the spleen, LNs, and thymus. (A) Representative dot plots show the gating strategy for cDC1 (CD11chiCD317−CD11b−CD8+), cDC2 (CD11chiCD317−CD11b+CD8−), and pDC (CD11cloCD317+B220+) identification in the spleens from Cxcr4+/1013 (+/1013) and littermate Cxcr4+/+ (WT) mice. The percentages of cDCs, CD11cloCD317+ cells, and pDCs among singlets and cDC1 and cDC2 among DCs are indicated. (B-C) Spleen cDCs and pDCs frequencies (B) and numbers (C) were determined by flow cytometry (n = 17 mice/group from 7 experiments). (D,G) Representative dot plots showing pDC gating in inguinal LNs (D) and the thymus (G) from +/1013 and WT mice. (E-F) Resident cDC (resDC, CD11chiCD317−MHCII+) and pDC (CD11cloCD317+B220+) frequencies (E) and numbers (F) in inguinal LNs from +/1013 and WT mice (n = 12-13 mice/group from 5 experiments). (H-I) cDC (CD11chiCD317−) and pDC (CD11cloCD317+B220+) frequencies (H) and numbers (I) in thymuses from +/1013 and WT mice (n = 7-8 mice/group from 3 experiments). Bar graphs show the mean ± SEM. Mice had a C57BL/6J genetic background. Statistical analysis was performed using the 2-tailed, unpaired Mann-Whitney test. *P < .05, **P < .01, ***P < .001.
CXCR4 dysfunction limits pDC egress from the BM while preserving their differentiation
We analyzed BM from Cxcr4+/1013 and control mice to determine whether the reduced peripheral pDC+/1013 number resulted from a production defect. The frequency of pDC+/1013 was higher, although their total number remained similar to that of pDCWT (Figure 3A-B) because of low BM cellularity (supplemental Figure 1E). Pre-cDC+/1013 frequencies and numbers were preserved. Thus, WS-associated CXCR4 dysfunction does not hamper pDC nor pre-cDC differentiation. CXCR4 blockade upon injection of AMD3100 led to peripheral mobilization of pDC+/1013 and normalization of their circulating counts (Figure 3C), consistent with these cells being trapped in the BM. Medullar pDC+/1013 and pDCWT expressed similar levels of CXCR4 (supplemental Figure 4A), ruling out pDC+/1013 retention resulting from increased CXCR4 cell-membrane expression. Further analysis of Ki67 and cleaved caspase 3 expression (supplemental Figure 4B-C) support the conclusion that pDC+/1013 divide less and are more prone to apoptosis than pDCWT, likely because of their medullar retention.
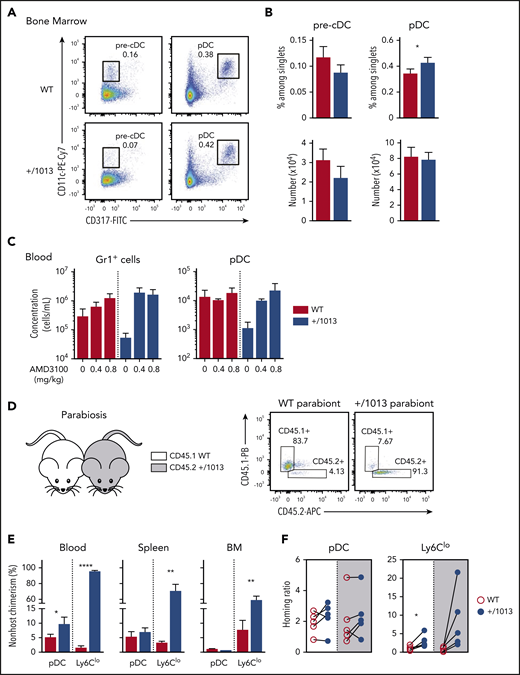
The peripheral pDC defect in Cxcr4+/1013 mice results from impaired CXCR4-dependent pDC egress from BM. (A) Representative dot plots showing the gating strategy to identify pre-cDCs (CD11cloCD317− gated on live CD4−CD8−B220−CD11b+MHC2−/int cells) and pDCs (CD11cloCD317+ among live B220+ cells) in the BM of +/1013 and WT mice. (B) Pre-cDC and pDC frequencies (top bar graphs) and numbers (bottom bar graphs) in the BM of +/1013 and WT mice (means ± SEM, n = 7-8 mice from 4 experiments for pre-cDC, n = 17 mice/group from 6 experiments for pDC frequency, n = 10-11 mice from 4 experiments for pDC number). (C) Numbers of blood pDCs and Gr1+ cells assessed 2 hours after intraperitoneal injection of AMD3100 or phosphate-buffered saline (mean ± SEM, n = 3 mice/group, 1 representative experiment of 2). (D-F) Parabiosis experiments were performed with CD45.1 Cxcr4+/+ (WT, left parabiont) and CD45.2 Cxcr4+/1013 (+/1013, right parabiont) mice. Blood, spleen, and BM were analyzed 8 weeks later. (D, right) Representative dot plots showing CD45.1 and CD45.2 expression in blood pDCs from WT and +/1013 parabionts. (E) Nonhost chimerism in the blood, spleen, and BM from each parabiont. pDC was defined as live CD3−Ly6G−CD11b−CD115−CD11cloCD317+Ly6C+B220+ cells and Ly6Clo monocytes as live CD3−B220−Ly6G−CD115+CD11b+CD317−Ly6Clo cells. (F) Spleen/blood ratios of WT and Cxcr4+/1013 pDCs and Ly6Clo monocytes calculated for the spleen of WT (white background) and Cxcr4+/1013 (gray background) parabionts (mean ± SEM, n = 5 pairs of mice). Mice had a C57BL/6J genetic background. Statistical analysis was performed using the paired t test. *P < .05, **P < .01, ****P < .001.
The peripheral pDC defect in Cxcr4+/1013 mice results from impaired CXCR4-dependent pDC egress from BM. (A) Representative dot plots showing the gating strategy to identify pre-cDCs (CD11cloCD317− gated on live CD4−CD8−B220−CD11b+MHC2−/int cells) and pDCs (CD11cloCD317+ among live B220+ cells) in the BM of +/1013 and WT mice. (B) Pre-cDC and pDC frequencies (top bar graphs) and numbers (bottom bar graphs) in the BM of +/1013 and WT mice (means ± SEM, n = 7-8 mice from 4 experiments for pre-cDC, n = 17 mice/group from 6 experiments for pDC frequency, n = 10-11 mice from 4 experiments for pDC number). (C) Numbers of blood pDCs and Gr1+ cells assessed 2 hours after intraperitoneal injection of AMD3100 or phosphate-buffered saline (mean ± SEM, n = 3 mice/group, 1 representative experiment of 2). (D-F) Parabiosis experiments were performed with CD45.1 Cxcr4+/+ (WT, left parabiont) and CD45.2 Cxcr4+/1013 (+/1013, right parabiont) mice. Blood, spleen, and BM were analyzed 8 weeks later. (D, right) Representative dot plots showing CD45.1 and CD45.2 expression in blood pDCs from WT and +/1013 parabionts. (E) Nonhost chimerism in the blood, spleen, and BM from each parabiont. pDC was defined as live CD3−Ly6G−CD11b−CD115−CD11cloCD317+Ly6C+B220+ cells and Ly6Clo monocytes as live CD3−B220−Ly6G−CD115+CD11b+CD317−Ly6Clo cells. (F) Spleen/blood ratios of WT and Cxcr4+/1013 pDCs and Ly6Clo monocytes calculated for the spleen of WT (white background) and Cxcr4+/1013 (gray background) parabionts (mean ± SEM, n = 5 pairs of mice). Mice had a C57BL/6J genetic background. Statistical analysis was performed using the paired t test. *P < .05, **P < .01, ****P < .001.
We further investigated the contribution of CXCR4 to pDC trafficking and homing to and residency in LO by establishing parabiosis between CD45.2 Cxcr4+/1013 and CD45.1 control mice (Figure 3D-F). Although nonhost chimerism was limited for blood pDCs (Figure 3E), in accordance with their short half-life in circulation,57,58 it was twofold higher in Cxcr4+/1013 parabionts than in WT ones. This difference likely reflects the reduced number of circulating pDC+/1013 (Figure 3E). In contrast, such a relative increase of pDCWT over pDC+/1013 was observed neither in the BM nor spleens of Cxcr4+/1013 parabionts, suggesting that CXCR4 dysfunction may affect pDC entry into these organs. We thus calculated spleen/blood ratios,50 which allowed normalization for cell chimerism. These ratios were similar for pDC+/1013 and pDCWT, regardless of the spleen genetic background, suggesting similar entry and residency of these cells in this LO. This contrasted with nonclassical Ly6Clo monocyte ratios (Figure 3F) whose trafficking was affected by CXCR4 dysfunction as reported.50 Thus, the quantitative defects in peripheral pDC+/1013 likely result from their reduced egress from the BM, as described for Cxcr4+/1013 BM neutrophils,45 confirming the reported role of CXCR4 in pDC egress from BM.31 Meanwhile, pDC differentiation, entry, and residency in the spleen are preserved.
pDC+/1013 display efficient innate functions
Most WHIM patients suffer from severe HPV-associated pathogenesis, suggesting impaired antiviral responses. We thus investigated the impact of CXCR4 dysfunction on pDC functions. pDC+/1013 tended to produce less IFN-α than pDCWT upon Toll-like receptor 9 (TLR9) engagement by CpG-ODN (Figure 4A, left). However, they also survived less in culture (Figure 4A, right) and were more prone to apoptosis than pDCWT (Figure 4B). In addition, pDC-like BMDC+/1013 and BMDCWT produced similar levels of IFN-α upon TLR9 engagement (Figure 4C). Overall, these experiments suggest that pDC+/1013 display preserved IFN-α responses.
![CXCR4 dysfunction does not alter pDC function. (A) IFN-α secretion (left) by splenic pDC from +/1013 and WT mice stimulated with CpG-ODN (CpG) or not (−) for 18 hours. pDC numbers (right) at the end of the culture. Mean ± SEM, n = 5-6 per group, cumulative data from 2 experiments. (B) Cleaved Caspase 3 levels in freshly isolated spleen pDCs (live CD11cintCD317+ cells) from +/1013 and WT mice. Mean ± SEM, n = 9 per group, cumulative data from 3 experiments. (C) IFN-α secretion by Flt3-l-derived pDCs from +/1013 and WT mice stimulated with CpG-ODN for 18 hours. Mean ± SEM, n = 2 per group, cumulative data from 2 experiments. (D-J) +/1013 and WT mice were injected with CpG-DOTAP (CpG) or DOTAP only (vehicle [veh]). (D) Representative dot plot of the CD45+ cells recovered in the peritoneal lavage 24 hours after treatment. (E) Fold increase in the number of cells recovered in the peritoneal lavage, the reference being the WT veh mice. (F) Number of pDCs (live CD45+CD11b−F4/80−CD11cloLy6C+CD317+B220+ cells), resident macrophages (live CD45+CD11bhiF4/80hiLy6G−Ly6Cint to hi cells), and inflammatory myeloid cells (live Infl. myeloid, CD45+B220−CD11b+F4/80lo to +Ly6Chi cells) in the peritoneal lavage. (G) Serum cytokine levels at different timepoints. (H-I) Surface expression of MHC2 (H) and CD86 (I) by splenic pDC (CD11cintCD317+B220+). Representative histograms (left) and the fold increase in the MFI relative to WT veh mice (right) are shown. The gray histogram corresponds to isotype control. (J) Surface expression of CD69 by splenic NK cells (live CD3−NK1.1+CD49b+ cells). Representative dot plots (left), the percentage of CD69+ NK cells (middle), and CD69 MFI (right) are shown.) Mean ± SEM, n = 5-6 per group, cumulative data from 3 experiments (D-J). Mice had a C57BL/6J genetic background. Statistical analysis was performed using the 2-tailed unpaired Mann-Whitney test to compare veh- and CpG-treated mice (#P < .05) or the WT and +/1013 groups (*P < .05).](https://ash.silverchair-cdn.com/ash/content_public/journal/blood/137/20/10.1182_blood.2020006675/2/m_bloodbld2020006675f4.png?Expires=1767710807&Signature=bporYdmLVEhZ2~sbhNRic25M2BvW335kqC8nzgYm-~rqHEOZxjqyVXgPByqlz013-Tk~onJU3dkBzTbt0A41t3Rv16CYUdlnH2TgWJ5Cg6O1E1so4XmXZgRygteiUdbiAym4cpLtsPAxLpQ8Exb-RgCS03u0opGToU7MIufFPV5w7JD59daKQCjxhTKfy43qhCnnFOumxVjW7h3F6Sq8zZqizzkLIAxs2RNi71KWXUqX0HiU9WzdnG68jv4E66~Twp-DaPNncqmIURZ-TgsQIM2y8AKnP~wxXsvMpW6z7syAGGJ9nzVI1IOBTOdDE0h~SrDmzkoVniS-upwPQbnz7w__&Key-Pair-Id=APKAIE5G5CRDK6RD3PGA)
CXCR4 dysfunction does not alter pDC function. (A) IFN-α secretion (left) by splenic pDC from +/1013 and WT mice stimulated with CpG-ODN (CpG) or not (−) for 18 hours. pDC numbers (right) at the end of the culture. Mean ± SEM, n = 5-6 per group, cumulative data from 2 experiments. (B) Cleaved Caspase 3 levels in freshly isolated spleen pDCs (live CD11cintCD317+ cells) from +/1013 and WT mice. Mean ± SEM, n = 9 per group, cumulative data from 3 experiments. (C) IFN-α secretion by Flt3-l-derived pDCs from +/1013 and WT mice stimulated with CpG-ODN for 18 hours. Mean ± SEM, n = 2 per group, cumulative data from 2 experiments. (D-J) +/1013 and WT mice were injected with CpG-DOTAP (CpG) or DOTAP only (vehicle [veh]). (D) Representative dot plot of the CD45+ cells recovered in the peritoneal lavage 24 hours after treatment. (E) Fold increase in the number of cells recovered in the peritoneal lavage, the reference being the WT veh mice. (F) Number of pDCs (live CD45+CD11b−F4/80−CD11cloLy6C+CD317+B220+ cells), resident macrophages (live CD45+CD11bhiF4/80hiLy6G−Ly6Cint to hi cells), and inflammatory myeloid cells (live Infl. myeloid, CD45+B220−CD11b+F4/80lo to +Ly6Chi cells) in the peritoneal lavage. (G) Serum cytokine levels at different timepoints. (H-I) Surface expression of MHC2 (H) and CD86 (I) by splenic pDC (CD11cintCD317+B220+). Representative histograms (left) and the fold increase in the MFI relative to WT veh mice (right) are shown. The gray histogram corresponds to isotype control. (J) Surface expression of CD69 by splenic NK cells (live CD3−NK1.1+CD49b+ cells). Representative dot plots (left), the percentage of CD69+ NK cells (middle), and CD69 MFI (right) are shown.) Mean ± SEM, n = 5-6 per group, cumulative data from 3 experiments (D-J). Mice had a C57BL/6J genetic background. Statistical analysis was performed using the 2-tailed unpaired Mann-Whitney test to compare veh- and CpG-treated mice (#P < .05) or the WT and +/1013 groups (*P < .05).
CXCR4 dysfunction does not alter pDC function. (A) IFN-α secretion (left) by splenic pDC from +/1013 and WT mice stimulated with CpG-ODN (CpG) or not (−) for 18 hours. pDC numbers (right) at the end of the culture. Mean ± SEM, n = 5-6 per group, cumulative data from 2 experiments. (B) Cleaved Caspase 3 levels in freshly isolated spleen pDCs (live CD11cintCD317+ cells) from +/1013 and WT mice. Mean ± SEM, n = 9 per group, cumulative data from 3 experiments. (C) IFN-α secretion by Flt3-l-derived pDCs from +/1013 and WT mice stimulated with CpG-ODN for 18 hours. Mean ± SEM, n = 2 per group, cumulative data from 2 experiments. (D-J) +/1013 and WT mice were injected with CpG-DOTAP (CpG) or DOTAP only (vehicle [veh]). (D) Representative dot plot of the CD45+ cells recovered in the peritoneal lavage 24 hours after treatment. (E) Fold increase in the number of cells recovered in the peritoneal lavage, the reference being the WT veh mice. (F) Number of pDCs (live CD45+CD11b−F4/80−CD11cloLy6C+CD317+B220+ cells), resident macrophages (live CD45+CD11bhiF4/80hiLy6G−Ly6Cint to hi cells), and inflammatory myeloid cells (live Infl. myeloid, CD45+B220−CD11b+F4/80lo to +Ly6Chi cells) in the peritoneal lavage. (G) Serum cytokine levels at different timepoints. (H-I) Surface expression of MHC2 (H) and CD86 (I) by splenic pDC (CD11cintCD317+B220+). Representative histograms (left) and the fold increase in the MFI relative to WT veh mice (right) are shown. The gray histogram corresponds to isotype control. (J) Surface expression of CD69 by splenic NK cells (live CD3−NK1.1+CD49b+ cells). Representative dot plots (left), the percentage of CD69+ NK cells (middle), and CD69 MFI (right) are shown.) Mean ± SEM, n = 5-6 per group, cumulative data from 3 experiments (D-J). Mice had a C57BL/6J genetic background. Statistical analysis was performed using the 2-tailed unpaired Mann-Whitney test to compare veh- and CpG-treated mice (#P < .05) or the WT and +/1013 groups (*P < .05).
We next tested the impact of CXCR4 dysfunction on the capacity of pDCs to initiate innate immune responses in a model of CpG-induced inflammation, in which pDCs are instrumental for myeloid cell recruitment to the peritoneum and natural killer (NK) cell activation.56 Upon CpG-ODN injection, both Cxcr4+/1013 mice and their controls showed increased cellularity and marked changes in the type of cells recovered from peritoneal lavages relative to vehicle (veh)-treated mice (Figure 4D-E). Although threefold fewer pDCs were present in the peritoneum of Cxcr4+/1013 than WT CpG-injected mice, the recruitment of inflammatory myeloid cells and the decrease in resident macrophages were similar in these mice (Figure 4F). Accordingly, the concentrations of inflammatory chemokines in the sera of Cxcr4+/1013 and WT CpG-injected mice increased similarly (Figure 4G). Furthermore, splenic pDCs from these mice overexpressed MHC2 and CD86 at equivalent levels (Figure 4H-I). Moreover, CD69+-activated NK cells were also detected at similar frequencies in these mice (Figure 4J). Thus, despite a quantitative defect, pDC+/1013 were recruited to the peritoneum, activated, and initiated systemic inflammation upon TLR9 engagement in vivo, as did pDCWT, supporting that CXCR4 desensitization is dispensable for their innate functions.
CXCR4 dysfunction promotes skin inflammation without affecting DC activation
Given the skin-associated symptoms reported in WHIM patients, we next investigated the impact of CXCR4 dysfunction on skin DC subsets and LCs. We used HPV mice47-49 to model HPV-induced dysplasia and chronic inflammation. The increase in flank epidermal thickness observed in HPV mice was worsened in HPV+/1013 mice (Figure 5A) and associated with higher numbers of dermal CD3dim T cells than in HPV mice (supplemental Figure 5A-C),47,52 suggesting greater inflammation in the skin of HPV+/1013 than HPV mice. LC density was diminished in the epidermal sheets in the context of HPV-induced dysplasia and their round morphology with short dendrites reflected their activation, independently of CXCR4 functioning (Figure 5B). We next analyzed DC subsets and LCs in skin cell suspensions (supplemental Figure 5D). pDCs were very rare in these suspensions, regardless of mouse genotype (not shown). HPV-induced dysplasia led to an accumulation of cDC subsets and LCs (Figure 5D). The frequency and number of cDC1, cDC2, and LCs among CD45+ cells did not differ according to CXCR4 genotype, although cDC1 and LCs tended to accumulate more in HPV+/1013 than HPV mice (Figure 5C-D). In the context of HPV, MHC2 expression was reduced in cDCs and LCs (Figure 5E-F, upper). CD80 expression was upregulated in cDC1, cDC2, and LCs from HPV and HPV+/1013 mice, as was that of CD86 in LCs and cDC1 (Figure 5E-F, middle and lower). Overall, these results show that the skin-DC compartment is generally preserved in the Cxcr4+/1013 context, despite the worsening of HPV-associated skin inflammation.
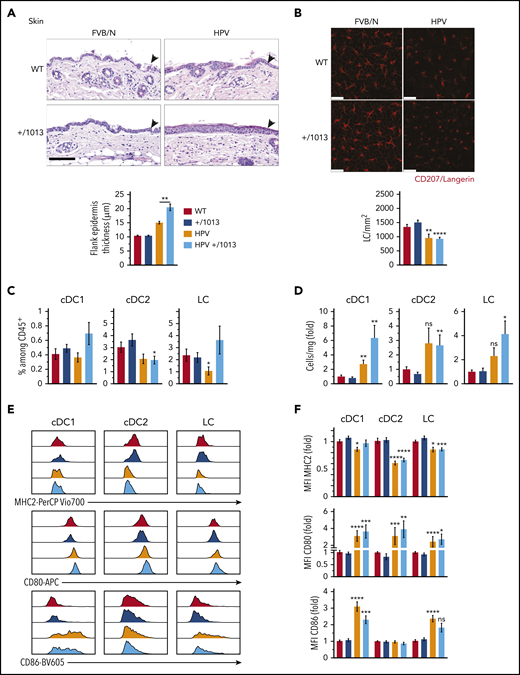
CXCR4 dysfunction promotes HPV-associated chronic inflammation while preserving DC subset activation. (A) Representative images for hematoxylin and eosin coloration of flank skin sections (upper). Epidermal (arrowheads) thickness was measured along the tissue, with at least 10 measurements per sample (lower, mean ± SEM, n = 6-8 mice). Scale bar, 100 µm. (B) Representative confocal microscopy images (top) from epidermal sheets stained for CD207 (red). CD207+ cells were counted in 5 fields per sample (n = 3 samples per genotype) and the cell density calculated (bottom). Scale bars, 25 µm. (C) Percentage of cDC1, cDC2, and LCs among CD45+ skin cells. (D) Number of cDC1, cDC2, and LC per milligram of skin as the fold change relative to WT (see supplemental Figure 5 for the gating strategy, mean ± SEM, n = 9-11 per group, cumulative data from 4 independent experiments). (E) Representative histograms showing the surface expression of MHC2, CD80, and CD86 by cDC1, cDC2, and LCs from WT, +/1013, HPV, and HPV+/1013 mice. (F) Bar graphs show the expression of MHC2, CD80, and CD86 as the fold change relative to WT (mean ± SEM, n = 10-12 mice/group for MHC2 from 5 experiments, n = 9-11 mice/group for CD86 from 4 experiments, n = 7-9 mice/group for CD80 from 3 experiments). Mice had an FVB/N genetic background. Statistical analysis was performed using the 2-tailed, unpaired Mann-Whitney test to compare HPV mice and HPV+/1013 genotypes (A, **P < .01) or HPV and HPV+/1013 mice to their non-HPV counterparts (B-E, *P < .05, **P < .01, ***P < .005, ****P < .001).
CXCR4 dysfunction promotes HPV-associated chronic inflammation while preserving DC subset activation. (A) Representative images for hematoxylin and eosin coloration of flank skin sections (upper). Epidermal (arrowheads) thickness was measured along the tissue, with at least 10 measurements per sample (lower, mean ± SEM, n = 6-8 mice). Scale bar, 100 µm. (B) Representative confocal microscopy images (top) from epidermal sheets stained for CD207 (red). CD207+ cells were counted in 5 fields per sample (n = 3 samples per genotype) and the cell density calculated (bottom). Scale bars, 25 µm. (C) Percentage of cDC1, cDC2, and LCs among CD45+ skin cells. (D) Number of cDC1, cDC2, and LC per milligram of skin as the fold change relative to WT (see supplemental Figure 5 for the gating strategy, mean ± SEM, n = 9-11 per group, cumulative data from 4 independent experiments). (E) Representative histograms showing the surface expression of MHC2, CD80, and CD86 by cDC1, cDC2, and LCs from WT, +/1013, HPV, and HPV+/1013 mice. (F) Bar graphs show the expression of MHC2, CD80, and CD86 as the fold change relative to WT (mean ± SEM, n = 10-12 mice/group for MHC2 from 5 experiments, n = 9-11 mice/group for CD86 from 4 experiments, n = 7-9 mice/group for CD80 from 3 experiments). Mice had an FVB/N genetic background. Statistical analysis was performed using the 2-tailed, unpaired Mann-Whitney test to compare HPV mice and HPV+/1013 genotypes (A, **P < .01) or HPV and HPV+/1013 mice to their non-HPV counterparts (B-E, *P < .05, **P < .01, ***P < .005, ****P < .001).
Skin DC and LC migration is impaired by CXCR4 dysfunction
We next assessed the impact of CXCR4 dysfunction on the migration of skin DCs to SDLNs (Figure 6A). Both the frequency and number of migDCs were lower in the SDLNs of Cxcr4+/1013 and HPV+/1013 mice than those of their control counterparts (Figure 6A-C). We performed FITC painting experiments to determine whether the reduction in migDC+/1013 numbers resulted from a migration defect (Figure 6D). The number of FITC+ migDC1 and migDC2+LC recovered from the SDLNs of Cxcr4+/1013 mice was reduced (Figure 6E), similarly to that of the FITC− DCs. This diminution was not associated with reduced CCR7 or CXCR4 expression (Figure 6F). We next tested whether CXCR4 blockade would restore Cxcr4+/1013 DC migration. AMD3100 injection reduced the migration of migDCWT to SDLNs, whereas no increase in migDC+/1013 migration could be observed (Figure 6G). Overall, these results show that dysfunctional CXCR4 signal termination impairs skin-DC migration to and/or their entry into SDLNs at steady state, as well as in acute and chronic inflammation.
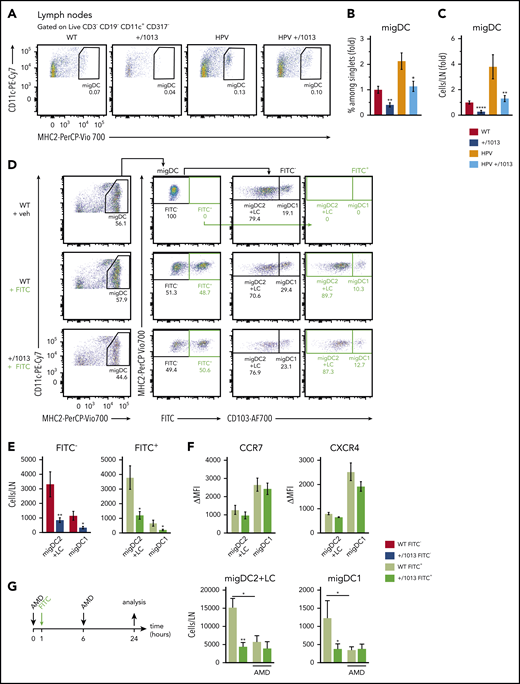
CXCR4 controls steady-state skin DC migration and in acute inflammation. (A) Representative dot plots showing migDCs (CD11c+MHC2hi gated on CD3−CD19−CD317−) in inguinal LNs from WT, +/1013, HPV, and HPV+/1013 mice. Percentages among parent cells are indicated. (B-C) Percentage among singlets (B) and number (C) of migDCs per LN as the fold change relative to WT values. (B-C) Mean ± SEM, n = 10-11 per group, cumulative data from four independent experiments. (D) Representative dot plots showing the gating strategy for identifying CD103+ migDC1 and CD103− migDC2+LC subsets in SDLNs from mice 24 hours after FITC treatment. (E) Number of FITC-negative (FITC−) and FITC-positive (FITC+) migDCs in skin-draining LNs from FITC-treated mice. (F) Membrane expression of CCR7 (left) and CXCR4 (right) by FITC+ migDC1 and migDC2+LC from WT and +/1013 mice. (G) Left, experimental design: mice received AMD3100 or PBS at t = 0 and 6 hours. FITC was applied at t = 1 hour. Right, number of FITC+ migDC1 and migDC2+LC in inguinal LNs form mice treated or not with AMD3100 (AMD). Mean ± SEM, n = 8-9 mice/group, cumulative data from 2 independent experiments (D-F). Mean ± SEM, n = 6-8 mice/group, cumulative data from 3 independent experiments (G). Mice had an FVB/N genetic background. Statistical analysis was performed using the 2-tailed, unpaired Mann-Whitney test to compare WT and +/1013 mice. *P < .05, **P < .01.
CXCR4 controls steady-state skin DC migration and in acute inflammation. (A) Representative dot plots showing migDCs (CD11c+MHC2hi gated on CD3−CD19−CD317−) in inguinal LNs from WT, +/1013, HPV, and HPV+/1013 mice. Percentages among parent cells are indicated. (B-C) Percentage among singlets (B) and number (C) of migDCs per LN as the fold change relative to WT values. (B-C) Mean ± SEM, n = 10-11 per group, cumulative data from four independent experiments. (D) Representative dot plots showing the gating strategy for identifying CD103+ migDC1 and CD103− migDC2+LC subsets in SDLNs from mice 24 hours after FITC treatment. (E) Number of FITC-negative (FITC−) and FITC-positive (FITC+) migDCs in skin-draining LNs from FITC-treated mice. (F) Membrane expression of CCR7 (left) and CXCR4 (right) by FITC+ migDC1 and migDC2+LC from WT and +/1013 mice. (G) Left, experimental design: mice received AMD3100 or PBS at t = 0 and 6 hours. FITC was applied at t = 1 hour. Right, number of FITC+ migDC1 and migDC2+LC in inguinal LNs form mice treated or not with AMD3100 (AMD). Mean ± SEM, n = 8-9 mice/group, cumulative data from 2 independent experiments (D-F). Mean ± SEM, n = 6-8 mice/group, cumulative data from 3 independent experiments (G). Mice had an FVB/N genetic background. Statistical analysis was performed using the 2-tailed, unpaired Mann-Whitney test to compare WT and +/1013 mice. *P < .05, **P < .01.
CXCR4 dysfunction fosters the arrival of highly activated migDCs to SDLNs
We further investigated the role of CXCR4 in the activation of migDCs. MigDCs from HPV mice (migDCHPV) showed lower MHC2 levels than migDCWT (Figure 7A-B), consistent with previous reports.59,60 ResDCHPV and pDCHPV also showed lower MHC2 levels than their controls (Figure 7B-C), highlighting a systemic effect of HPV-induced inflammation on DC-subset activation. In contrast, MHC2 levels were not decreased in migDCs from HPV+/1013 mice (migDCHPV+/1013), resDCHPV+/1013, and pDCHPV+/1013. Moreover, migDCHPV+/1013 showed an activated phenotype, with higher levels of CD80 and CD86 than migDCHPV. Such an increase in costimulatory molecule expression, as well as that of CCR7, affected both migDC1 and migDC2+LC, although it was statistically significant only for migDC1 (Figure 7D). Remarkably, this pattern was also found in migDC+/1013, suggesting that it did not result from the increased skin inflammation observed in HPV+/1013 mice (supplemental Figure 5). This prompted us to investigate whether DC+/1013 were intrinsically more prone to activation than DCWT. cDC1-like and cDC2-like BMDC+/1013 and BMDCWT were similarly activated by TLR4 and TLR8 agonists, whether or not CXCL12 was added to the culture medium (Figure 7E). Overall, these experiments suggest that the increased activation of migDC+/1013 does not result from an intrinsically higher capacity to become activated, but rather from their altered migration dynamics.
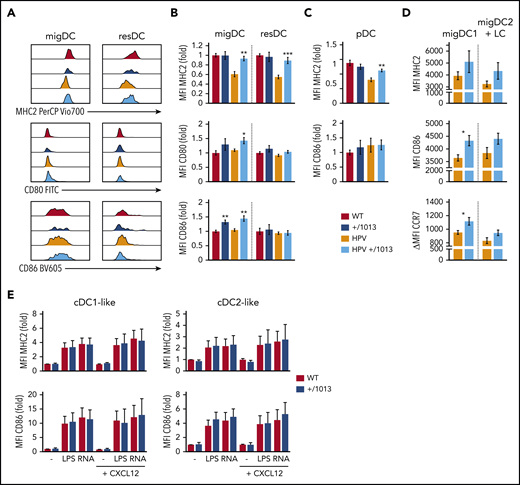
CXCR4 dysfunction promotes steady-state DC activation and in the context of HPV-associated inflammation. (A) Representative histograms showing the levels of MHC2, CD80, and CD86 surface expression by migDCs and resDCs (defined as CD11c+MHC2hi and CD11chiMHC2+ gated on live CD3−CD19−CD317− cells, respectively) recovered from the SDLNs of WT, +/1013, HPV, and HPV+/1013 mice. (B) Bar graphs showing the surface expression of MHC2, CD80, and CD86 for migDCs and resDCs as the fold change relative to WT. (C) Bar graphs showing the expression of MHC2 and CD86 in pDCs as the fold change relative to WT. Mean ± SEM, n = 8-11 mice/group from 4 experiments for MHC2, n = 8 mice/group from 3 experiments for CD86, n = 6 mice/group from 2 experiments for CD80 (B-C). (D) Bar graphs showing the surface expression of MHC2, CD86, and CCR7 for migDC1 and migDC2+LC from HPV and HPV+/1013 mice (mean ± SEM, n = 6-7 per group from 1 of 2 representative experiments). (E) Bar graphs showing the surface expression of MHC2 and CD86 upon LPS or ssRNA stimulation for DC1-like and DC2-like BMDCs derived from WT and +/1013 mice. Results are expressed as the fold change relative to nontreated WT (mean ± SEM, cumulative data from n = 4 to 5 independent experiments). Mice had an FVB/N genetic background. Statistical analysis was performed using the 2-tailed, unpaired Mann-Whitney test to compare +/1013 mice to their WT counterparts. **P < .01, ***P < .005.
CXCR4 dysfunction promotes steady-state DC activation and in the context of HPV-associated inflammation. (A) Representative histograms showing the levels of MHC2, CD80, and CD86 surface expression by migDCs and resDCs (defined as CD11c+MHC2hi and CD11chiMHC2+ gated on live CD3−CD19−CD317− cells, respectively) recovered from the SDLNs of WT, +/1013, HPV, and HPV+/1013 mice. (B) Bar graphs showing the surface expression of MHC2, CD80, and CD86 for migDCs and resDCs as the fold change relative to WT. (C) Bar graphs showing the expression of MHC2 and CD86 in pDCs as the fold change relative to WT. Mean ± SEM, n = 8-11 mice/group from 4 experiments for MHC2, n = 8 mice/group from 3 experiments for CD86, n = 6 mice/group from 2 experiments for CD80 (B-C). (D) Bar graphs showing the surface expression of MHC2, CD86, and CCR7 for migDC1 and migDC2+LC from HPV and HPV+/1013 mice (mean ± SEM, n = 6-7 per group from 1 of 2 representative experiments). (E) Bar graphs showing the surface expression of MHC2 and CD86 upon LPS or ssRNA stimulation for DC1-like and DC2-like BMDCs derived from WT and +/1013 mice. Results are expressed as the fold change relative to nontreated WT (mean ± SEM, cumulative data from n = 4 to 5 independent experiments). Mice had an FVB/N genetic background. Statistical analysis was performed using the 2-tailed, unpaired Mann-Whitney test to compare +/1013 mice to their WT counterparts. **P < .01, ***P < .005.
Discussion
We used patient-derived CXCR4 dysfunction, associated with a high susceptibility to HPV pathogenesis, to investigate the role of CXCR4 in DC biology. Our results identify prominent and subset-specific effects of CXCR4 signal termination on DC distribution and function that underpin adequate immune homeostasis and function. They suggest that the CXCL12/CXCR4 signaling axis sets a threshold for both pDC egress from the BM and LC/cDC migration from the dermis to SDLNs, which has a profound impact on migDC activation.
We provide the demonstration that CXCR4 activity regulates the abundance of peripheral pDCs, both in humans and mice, presumably through the control of pDC egress from the BM, while not altering their entry into the spleen nor residency. We rule out the hypothesis that reduced peripheral pDC+/1013 results from a defect in their production because their frequency was higher and their number preserved in the BM. Nevertheless, a bias toward myeloid-derived pDC differentiation7 could explain the lower SiglecH levels in splenic pDC+/1013 than pDCWT. Chopin et al31 reported that the level of CXCR4 expression by pDCs regulates their retention in the BM. Our study shows that the WS-associated CXCR4 dysfunction mimics the consequences of defective CXCR4 downregulation,31 thus highlighting the critical importance of the proper regulation of the CXCR4 signaling in pDC trafficking. In a model of CpG-induced inflammation that strictly depends on pDC,56 we demonstrate that pDC+/1013 can be mobilized and activated in vivo, induce inflammatory responses, and activate NK cells to the same extent as pDCWT. In addition, pDC+/1013 produced IFN-α as efficiently as pDCWT upon TLR9 engagement. Thus, our results do not support intrinsic pDC dysfunction as the mechanism contributing to WHIM patients’ susceptibility to infectious diseases, although reduced pDC numbers early in the course of infection may delay the mounting of an immune response.
Although our report is in accordance with previous findings of Tassone et al concerning the reduction of peripheral pDCs, it contradicts their conclusion that circulating cDC frequency and number are reduced in WHIM patients.44 Indeed, we show the frequency to be elevated for cDC1 and normal for cDC2 in WHIM patients relative to healthy donors. cDC numbers were preserved as well. Different factors might explain this discrepancy. First, we used an antibody panel including anti-CD11c, anti-HLA-DR, anti-CD1c, and anti-CD141 antibodies to gate on cDC subsets, whereas Tassone et al used anti-CD4 and anti-CD1c antibodies to identify cDCs. Second, we have worked on whole blood and not on isolated peripheral blood mononuclear cells. Moreover, we cannot rule out that differences in patient treatment might affect cDC concentrations. Supporting our data in humans, the cDC frequency was elevated in the spleens of Cxcr4+/1013 mice. These results are consistent with preserved myeloid differentiation and may also partially result from the local extramedullary hematopoiesis reported for these mice.46 Our data further show that the number, but not frequency, of circulating pre-cDCs was reduced by CXCR4 dysfunction, consistent with the role reported for CXCR4 in their medullar retention.32,61 Nevertheless, the resDC compartment was globally preserved in both lymphoid and nonlymphoid tissues from Cxcr4+/1013 mice, in support of efficient local differentiation of pre-cDCs and, in the skin, as a possible result of cDC entrapment and accumulation.
WHIM patients suffer from severe HPV-induced warts.38,41-43 We show that WS-associated CXCR4 dysfunction is associated with a marked reduction in DC and LC migration from the skin to SDLNs. Remarkably, CXCR4 blockade in control mice similarly induced a reduction in skin DC migration. Thus, although CXCR4 promotes skin DC migration to SDLNs as previously reported by Kabashima et al,26 the WS-associated gain of CXCR4 function completely abrogates this pro-migratory effect. Our interpretation of this apparently counterintuitive migration defect in Cxcr4+/1013 mice is that DCs and LCs might be trapped in the dermis, where CXCL12 is produced,23-28 despite their expression of CCR7, and as a consequence of a possibly skewed balance between several chemotactic signals. Indeed, Ricart et al reported that in vitro countergradients of CXCL12 and CCL19/CCL21 can result in the homing of DCs to a central zone, where chemokine-derived signals are balanced.62 Moreover, the levels of CXCL12 were shown to be higher in the dermis of Cxcr4+/1013 mice than that of their controls,27 suggesting that DC+/1013 entrapment could arise from the combined effect of high CXCL12 quantities in the dermis and exacerbated CXCR4-dependent signaling. Because CXCL12 is expressed by endothelial cells from the lymphatics,26 migDC+/1013 may also be trapped in these vessels on their way to LNs. Our results show that CCR7 expression is upregulated in migDC+/1013 compared with migDCWT in the HPV context, supporting the hypothesis that only the DCs that were highly responsive to CCL19/CCL21 signals could escape CXCL12 attraction in Cxcr4+/1013 mice. That no CCR7 upregulation was detected in migDC+/1013 in FITC-painting experiments suggests the contribution of other drivers of DC migration during acute inflammation. Importantly, CXCR4 blockade did not rescue migDC+/1013 migration to SDLNs. This might reflect sequential roles for CXCR4 and CCR7 in guiding skin DC migration, or, alternatively, an abrogation of the cooperativity between CXCR4 and CCR7 that was previously reported in pDCs and T cells.33,63 Overall, our data suggest that proper CXCR4 regulation controls skin immune homeostasis by setting up a threshold for cDC and LC migration from the dermis to SDLNs.
CXCR4 dysfunction is associated with profound alteration of the activation of the DC subsets and LCs recovered from SDLNs, with specific features in steady-state and chronic HPV-induced inflammation. First, CXCR4 dysfunction results in increased expression of costimulatory molecules by SDLN migDC, but not resDC or pDC. This occurs both at steady-state and in the context of HPV-induced inflammation, ruling out the possibility that the high activation level of migDC is simply because of the increased inflammation observed in the skin of HPV+/1013 mice. Upon in vitro stimulation, BMDC+/1013 were similarly activated as BMDCWT, suggesting that the increased activation of migDC+/1013 in SDLNs is not intrinsically programmed. Accordingly, the skin DC and LC phenotype was similar in HPV and HPV+/1013 mice. Thus, we propose that the high expression of costimulatory molecules by migDC+/1013 results from the stringent selection of DCs exiting the skin, meaning that only the most activated cells would leave the CXCL12-enriched dermis and reach the LNs. Second, CXCR4 dysfunction also markedly affects MHC2 expression by SDLN DCs in the context of chronic inflammation. Indeed, HPV-induced inflammation was associated with marked downregulation of MHC2 expression by skin DCs, as expected from previous studies,59,60 and independently of CXCR4 status. Such a tolerogenic state of DCs was reported to promote skin graft tolerance64 and could be a component of HPV immune-escape mechanisms. However, although MHC2 downregulation was also present in SDLN migDCHPV, resDCHPV, and pDCHPV, this pattern was not found in any of these subsets in HPV+/1013 mice. This absence of a tolerogenic profile affecting all LN DC subsets may be a distal consequence of exacerbated skin inflammation in HPV+/1013 mice. Such worsening of HPV-induced pathogenesis may arise from the cumulative effects of the mutant CXCR4 procarcinogenetic properties in keratinocytes65,66 and effect on stromal and immune cells, which could be further fostered by highly activated migDCs, promoting inflammation in HPV+/1013 mice.
Whether alterations in DC biology directly contribute to the high susceptibility of WHIM patients to HPV-induced pathogenesis remains to be investigated. One may hypothesize that inefficient priming of HPV-specific T-cell responses resulting from scarce numbers of DCs reaching the LN,67 combined with defective DC–T-cell interactions when T cells express WS-associated CXCR4 mutations,68 could hamper lesion control.69 Moreover, excessively activated DCs could be defective for CD8 T-cell preconditioning to become resident memory T cells70 involved in the local control of HPV,71 prime inappropriate T-cell responses, or fail in inducing tolerance.72,73 In support of the hypothesis that CXCR4 dysfunction can exacerbate immune responses, an increase in the generation of plasma cells was reported in the SLO of Cxcr4+/1013 mice, although it did not result in long-term protective humoral immunity.74 This raises the possibility that WHIM patients might paradoxically suffer from a lack of tolerance toward their skin microbiota, which includes the HPV virome,75,76 combined with defects in the mounting of anti-infectious immune responses. Further studies are needed to test these hypotheses, which may be conducted using mouse models of papillomavirus infection.77,78
Our work suggests that functional restoration of the DC compartment might have contributed to the spontaneous cure of HPV-induced warts reported in a WHIM patient who fortuitously lost the mutated CXCR4 allele in a single myeloid progenitor that eventually repopulated the myeloid repertoire.39 Indeed, the critical role of DCs in the priming of adaptive immune responses argues in favor of a contribution of the recovery of skin DC migration to the beneficial effects of this chromothriptic event.79 Our work also suggests that normalizing DC subsets trafficking, together with their activation, could be among the mechanisms accounting for the beneficial effects of the treatment of WHIM patients with AMD3100 combined with imiquimod on HPV-induced lesions and HPV cancer progression.42,80
In summary, our work provides mechanistic cues to explain the physiopathology of WS and unravels the profound impact of CXCR4-dependent signal termination on DC subsets migration dynamics and activation state.
For original data, please contact Géraldine Schlecht Louf (geraldine.schlecht-louf@universite-paris-saclay.fr).
The online version of this article contains a data supplement.
The publication costs of this article were defrayed in part by page charge payment. Therefore, and solely to indicate this fact, this article is hereby marked “advertisement” in accordance with 18 USC section 1734.
Acknowledgments
The authors thank Hélène Gary (Plaimmo, Unité Mixte de Service/Institut Paris-Sud d’Innovation Thérapeutique [UMS/IPSIT], Châtenay-Malabry, France), Sarah Mendez, Baptiste Lecomte (Animex, UMS/IPSIT), and Khadidiatou Kouyate (Laboratory “Inflammation, Microbiome and Immunosurveillance”) for excellent technical assistance. The authors also thank Floriane Meuris for backcrossing the Cxcr4+/1013 mice onto the FVB/N genetic background and are grateful to Matthieu Bertrand, Patrice Hémon, and Chloé Lourenço for performing preliminary experiments.
This work was supported by grants from the ERA-Net Infect-ERA “HPV-MOTIVA” (Agence Nationale de la Recherche [ANR]-15-IFEC-0004-01) (F.B. and G.S.L.), MSCA-ITN-2014-ETN/Marie Skłodowska-Curie Innovative Training Networks (ITN-ETN) “ONCOgenic Receptor Network of Excellence and Training” (H2020-MSCA Program, grant agreement 641833-ONCORNET) (F.B. and G.S.L.), the “Fondation ARC pour la Recherche sur le Cancer” (G.S.L.), LabEx LERMIT (Laboratory of Excellence in Research on Medication and Innovative Therapeutics) (ANR-10-LABX-33) under the program “Investissements d’Avenir” (ANR-11-IDEX-0003-01) (F.B. and G.S.L.), and ANR Projet de Recherche Collaborative (PRC) grant ANR-17-CE14-0019 (K.B.). C.G. was supported by the MSCA-ITN-2014-ETN/ITN-ETN “ONCOgenic Receptor Network of Excellence and Training” and the Fondation pour la Recherche Médicale (FDT201805005700). M.V. and J.C. were supported by a doctoral fellowship from the Doctoral School “Innovation Thérapeutique du Fondamental à l’Appliqué” (ED 569). M.V. was supported by the “Ligue contre le Cancer” (IP/SC-15956). N.P. was supported by a postdoctoral fellowship from the Région Ile de France (Domaine d'Intérêt Majeur [DIM] Maladies Infectieuses, Parasitaires et Nosocomiales Emergentes).
Authorship
Contribution: C.G. and M.V. designed and performed experiments, analyzed data, contributed to writing the manuscript, and worked as co-first authors, a position assigned on the basis of the number of experiements performed, analysed, and interpreted, as well as the contribution to the setting up of the techniques used; J.C., M.R., M.-L.A., N.P., M.E., and M.L. designed and performed experiments and analyzed data; V.M.-E. designed and performed experiments with patient samples, analyzed data, and contributed to writing the manuscript; F.M.-N. performed histology experiments and analyzed data; Y.B., F.S., and J.D. provided blood samples from healthy donors and WHIM patients and reviewed the manuscript; L.G.N. provided access to the parabiosis data and reviewed the manuscript; K.B. reviewed the manuscript; F.B. helped with the study design, data analysis, and writing of the manuscript and procured funding for the study; and G.S.-L. conceived and supervised the study, designed and performed experiments, analyzed data, procured funding for the study, and wrote the manuscript.
Conflict-of-interest disclosure: The authors declare no competing financial interests.
Correspondence: Géraldine Schlecht-Louf, INSERM UMR 996, 32 rue des Carnets, 92140 Clamart, France; e-mail: geraldine.schlecht-louf@universite-paris-saclay.fr.

This feature is available to Subscribers Only
Sign In or Create an Account Close Modal